

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 4, Number 6, June 2013, pages 420-423
Primary Cerebral Lymphoma Causing Remitting and Relapsing Neurological Symptoms
Thomas Stokera, e, Adam Younga, Tarik Massoudb, Rikie Patanic, d, Mark Manfordd
aSchool of Clinical Medicine, University of Cambridge, Addenbrooke’s Hospital, Cambridge, CB2 0SP, England, U.K.
bDepartment of Radiology, University of Cambridge, Addenbrooke’s Hospital, Cambridge, CB2 2QQ, England, U.K.
cAnne McLaren Laboratory for Regenerative Medicine, University of Cambridge, Cambridge, CB2 0SZ, England, U.K.
dDepartment of Neurology, Addenbrooke’s Hospital, Cambridge, CB2 0SP, England, U.K.
eCorresponding author: T. B. Stoker, School of Clinical Medicine, University of Cambridge, Addenbrooke’s Hospital, Cambridge, CB2 0SP, England, U.K.
Manuscript accepted for publication April 21, 2013
Short title: Remitting and Relapsing Neurological Symptoms
doi: https://doi.org/10.4021/jmc1159e
| Abstract | ▴Top |
Primary CNS lymphoma is a rare variant of non-Hodgkin’s lymphoma. Incidence has increased over the past 3 decades, but the optimum treatment protocol is yet to be established. We report a 44-year old man who presented with left-sided numbness. Magnetic resonance imaging of the brain revealed a right hemisphere ill-defined mass. Within four months the patient experienced a spontaneous near-full recovery. From this time the patient deteriorated with increasing left hemiparesis. After several months, the patient developed lethargy and dyspnoea, with episodes of epistaxis and per rectal bleeding. Full blood count revealed a pancytopaenia. Trephine biopsy demonstrated evidence of marrow involvement by diffuse large B-cell lymphoma. The patient was diagnosed with primary CNS lymphoma with systemic involvement. We conclude that the spontaneous remission of symptoms should not discourage a diagnosis of PCNSL if consistent with clinical and radiological findings.
Keywords: Primary central nervous system lymphoma; Diffuse large B cell lymphoma; Non-Hodgkin’s lymphoma
| Introduction | ▴Top |
Primary central nervous system lymphoma (PCNSL) is a rare variant of non-Hodgkin’s lymphoma, accounting for 3-5% of brain tumours [1]. Immunodeficiency is the only well-documented risk factor. Diffuse large B cell lymphoma (DLBCL) accounts for the vast majority of cases of PCNSL in immunocompetent patients. Although the overall incidence remains low, there has been an increase in the number of cases over the past 3 decades, particularly in immunocompetent individuals [2]. Owing to the low incidence of PCNSL, it has been difficult to establish the optimum treatment protocol for this disease [3].
| Case Report | ▴Top |
A 44-year-old man with no significant past medical history presented in April 2009 to his general practitioner with numbness in his left arm, and subsequently his left leg, causing him to limp with no significant discomfort. A brain MRI scan revealed a right hemisphere small ill-defined mass in the posterior centrum ovale and extending inferiorly towards the basal ganglia (Fig. 1A), which was considered to be inflammatory.
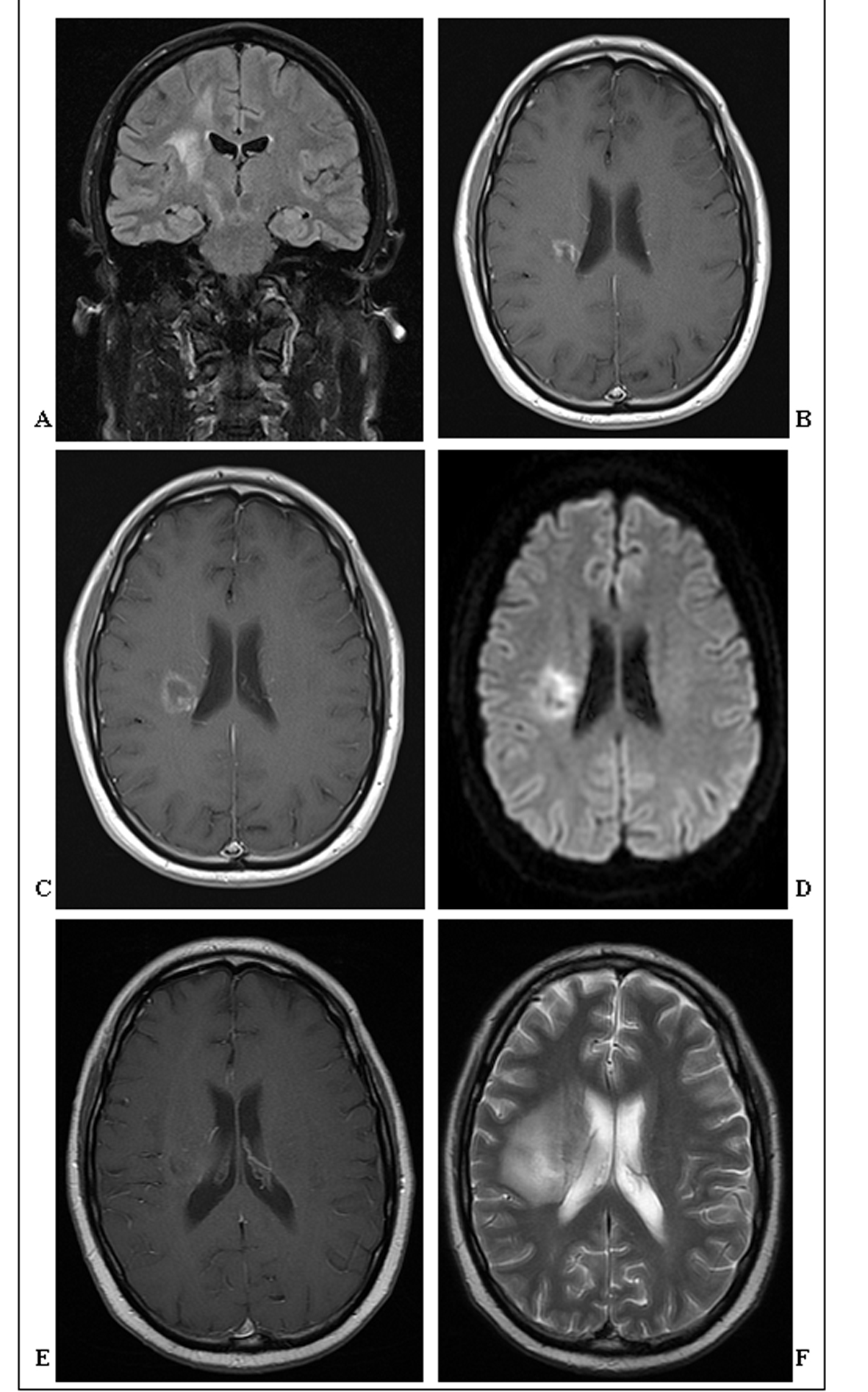 Click for large image | Figure 1. (A). Coronal FLAIR-MRI showing an ill-defined T2 hyperintense 1cm mass in the right posterior centrum semiovale extending inferiorly towards the basal ganglia. Surrounding vasogenic oedema extends throughout adjacent white matter. (B). Axial post contrast T1-weighted MRI showing greater definition of the same enhancing 1.4 cm mass in the right posterior centrum semiovale, now with some central cavitation. (C). Axial post contrast T1-weighted MRI showing further definition, increase in size (2 cm) and central cavitation of the mass in the right posterior centrum semiovale. There is irregular thick ring enhancement of the mass, but no significant mass effect. (D). Axial diffusion-weighted MRI showing restricted diffusion of the centrally cavitating mass in the right posterior centrum semiovale. (E). Axial post-contrast T1-weighted MRI showing poor definition and enhancement of the centrally cavitating mass in the right posterior centrum semiovale. (F). Axial T2-weighted MRI showing the extensive vasogenic oedema surrounding the hyperintense mass in the right posterior centrum semiovale. |
Within four months the patient experienced a spontaneous near-full recovery, enabling him to perform heavy exercise. An MRI scan at that time demonstrated that the ring-enhancing lesion had increased in size and developed central cavitation (Fig. 1B). From this time the patient began to clinically deteriorate with increasing left hemiparesis. On examination he had marked left upper motor neuron facial weakness, and power was 3/5 and 4/5 in the left arm and leg respectively.
In November 2009, the patient suddenly deteriorated, with an abrupt loss of sensation in the left arm and impairment of coordination in the left leg. There was no headache, dizziness, visual or hearing impairment. Clinical examination revealed fixed-flexion of the left arm, and brisk reflexes in the left lower limb accompanied by an up-going plantar reflex. Cranial nerve examination was normal, except for the previously reported left-sided weakness of the face, which was now accompanied by a loss of sensation. A third MRI scan demonstrated further progression, with increased size of the mass (2 cm), cavitation, and irregular thick ring enhancement (Fig. 1C).
At this time routine haematological examination was normal. Cerebrospinal fluid (CSF) examination was also within normal limits with an opening pressure of 12 cm (10 - 25 cm), 1.33 g/L protein (< 0.4g/L), 3.9 mmol/L glucose (2.8 - 4.4 mmol/L), normal cytology, negative bacterial culture and gram stain. No monoclonal or oligoclonal bands were detected and polymerase chain reaction for Toxoplasma, Cryptococcus, syphilis, hepatitis B and C viruses were negative. Anti-nuclear, anti-neutrophilic cytoplasmic, anti-mitochondrial, anti-smooth muscle, anti-liver-kidney microsomal antibodies were all negative. CSF angiotensin-converting enzyme however, was elevated at 115 U/L (< 1.2 U/L). Human immunodeficiency virus 1 and 2 antibodies were negative.
A CT scan of the chest, abdomen and pelvis was performed, which identified a mass on the lower pole of the right kidney measuring a maximum diameter of 6.6 cm. The possibility of renal cell carcinoma with cerebral metastases was considered, but renal biopsy revealed a diagnosis of an incidental oncocytoma.
Dexamethasone 6 mg TDS was initiated leading to a significant clinical improvement. Due to the development of intolerable side effects, the steroid dosage was gradually reduced to 2 mg BD, and azathioprine therapy was initiated. However, due to adverse effects (hepatic dysfunction), azathioprine was discontinued after one week.
Three months later the patient was admitted with weakness in the left side of the face, left arm (particularly the fingers) and left leg. On examination, there was marked weakness and hypertonia, and reflexes were brisk throughout the left side. Sensation was impaired in both the upper and lower left limbs, particularly light touch and pin-prick. The MRI at this stage revealed that the pre-existing lesion was of similar size but had become less discrete, and with less contrast enhancement but greater restricted diffusion (Fig. 1D).
In addition to the neurological features mentioned, the patient also complained of lethargy and dyspnoea on mild exertion together with episodes of epistaxis and per rectal bleeding. He did not report any weight loss or night sweats and there was no clinical evidence of lymphadenopathy. The full blood count demonstrated a pancytopaenia with 8.4g/dL haemoglobin (13 - 18 g/dL), 105.3 fL mean corpuscular volume (76 - 96 fL), 86 × 109/L platelets (150 - 400 × 109/L) and 4.8 × 109/L leukocytes (4 - 11 × 109/L).
In view of the patient’s deteriorating haematological profile bone marrow aspiration and trephine biopsy were performed. The bone marrow aspirate demonstrated abnormal mononuclear cells, the morphology favouring a haematopoietic process (for example, large cell lymphoma). There was also evidence of hemophagocytosis and erythroid hyperplasia, accounting for the anaemia. There was a grossly abnormal cellular infiltrate raising the possibility of haematological malignancy. Trephine biopsy revealed small clusters of cells in keeping with marrow involvement by DLBCL.
The patient was initiated on rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine and prednisolone therapy for treatment of PCNSL with bone marrow involvement. Two further brain MRI scans in May and July 2010 showed poorer definition of the lesion, which had remained constant in size, but with greater surrounding oedema (Fig. 1E, F). Unfortunately, the patient’s symptoms progressed to include visual impairment, and power on the left side deteriorated to 0/5 throughout. Treatment was complicated by several episodes of neutropenic sepsis, from one of which the patient died, two months later.
| Discussion | ▴Top |
This case demonstrates an unusual presentation of a rare, but increasingly encountered condition in immunocompetent hosts [3]. Owing to the relative rarity of PCNSL, it has proved difficult to study, and the optimum treatment strategy has yet to be determined [3].
Clinical manifestations of PCNSL include neuropsychiatric signs, raised intracranial pressure, seizures, ocular symptoms and other focal deficits [1, 4]. The so-called, ‘B symptoms’ include weight loss, fever and night sweats, and are rarely encountered at initial presentation [4].
Neuroimaging is important in the diagnosis of PCNSL, with contrast-enhanced MRI being the modality of choice [4]. Lesions may be multiple, and may resemble inflammatory and infectious diseases, as well as other neoplastic processes [1]. PCNSL masses have acquired the pseudonym of “ghost tumours”, as radiological changes may disappear, particularly after corticosteroid therapy [1]. Definitive diagnosis is achieved by stereotactic biopsy, although this is sometimes unnecessary owing to the presence of lymphomatous cells in the CSF [1]. Glucocortcoid therapy may obscure the histological diagnosis, so steroids should not be administered (pre-biopsy) if PCNSL is likely [5].
The development of effective chemotherapy regimens has meant that a proportion of patients may achieve remission. Relapse however, is a common occurrence, and only 20-30% of patients are cured [1]. The spontaneous remission of symptoms that occurred initially in this case is an extremely unusual occurrence, and may impose a significant diagnostic challenge in cases with PCNSL. Consequently, this patient’s symptoms were initially considered to be due to an inflammatory or demyelinating process, culminating in a delay in the initiation of appropriate cytotoxic therapy.
The diagnosis of PCNSL in the case described was not established until symptoms of bone marrow involvement had developed. The incidence of systemic involvement following PCNSL is controversial, with some studies suggesting that extensive staging is unnecessary [1]. However, investigation for occult systemic disease is increasingly undertaken, with full body CT scanning and bone marrow biopsy now recommended for staging of PCNSL [3]. Testicular ultrasonography may be performed in elderly males, as testicular lymphoma frequently disseminates to the brain [3].
Conclusion
The incidence of PCNSL in immunocompetent hosts is increasing. Spontaneous remission of symptoms is unusual, but can occur, and should not discourage a differential diagnosis of PCNSL if consistent with clinical and radiological findings. Extra-neural disease is increasingly recognised in PCNSL, and patients should undergo thorough staging to exclude systemic disease.
Declaration
The authors declare no competing interest.
| References | ▴Top |
- Sierra del Rio M, Rousseau A, Soussain C, Ricard D, Hoang-Xuan K. Primary CNS lymphoma in immunocompetent patients. Oncologist. 2009;14(5):526-539.
doi pubmed - Miller DC, Hochberg FH, Harris NL, Gruber ML, Louis DN, Cohen H. Pathology with clinical correlations of primary central nervous system non-Hodgkin's lymphoma. The Massachusetts General Hospital experience 1958-1989. Cancer. 1994;74(4):1383-1397.
doi - Abrey LE, Batchelor TT, Ferreri AJ, Gospodarowicz M, Pulczynski EJ, Zucca E, Smith JR, et al. Report of an international workshop to standardize baseline evaluation and response criteria for primary CNS lymphoma. J Clin Oncol. 2005;23(22):5034-5043.
doi pubmed - Gerstner ER, Batchelor TT. Primary central nervous system lymphoma. Arch Neurol. 2010;67(3):291-297.
doi pubmed - Weller M. Glucocorticoid treatment of primary CNS lymphoma. J Neurooncol. 1999;43(3):237-239.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.