

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 13, Number 3, March 2022, pages 129-134

Immunoglobulin G4-Associated Cholangitis: Two Case Reports and Clinical Use of Classic and Emerging International Diagnostic Criteria
Ricardo Jacaranda de Fariaa, b, e, Marcos Vasconcelos Carneiroc, Lucio Lucas Pereiraa, Raissa Dytzd, Gabriela dos Santosd
aInstituto de Gestao Estrategica de Saude do Distrito Federal (IGESDF), Hospital Universitario de Brasilia (HUB), Universidade de Brasilia (UnB), Asa Norte, Brasilia, DF 70840-901, Brazil
bSchool of Medicine, Centro Universitario de Brasilia (CEUB), SMHS Area Especial, Q1-Asa Sul, 70330-150, Brasilia, DF, Brazil
cIGESDF, Universidade Catolica de Brasilia (UCB), Campus Taguatinga, Taguatinga, 71966-700, Brasilia, DF, Brazil
dUniversidade Catolica de Brasilia (UCB), Brasilia, DF, Brazil
eCorresponding Author: Ricardo Jacaranda de Faria, Instituto de Gestao Estrategica de Saude do Distrito Federal (IGESDF), Hospital Universitario de Brasilia (HUB), Universidade de Brasilia (UnB), Asa Norte, Brasilia, DF 70840-901, Brazil
Manuscript submitted December 7, 2021, accepted February 21, 2022, published online March 5, 2022
Short title: Diagnosis of IgG4-Associated Cholangitis
doi: https://doi.org/10.14740/jmc3875
| Abstract | ▴Top |
Immunoglobulin G4-related disease is a multisystem disorder with unique gastrointestinal tract manifestations, often simulating neoplasms and other inflammatory conditions. Appropriate clinical suspicion and application of internationally validated criteria can assist in making the proper diagnosis. This article describes two cases of patients presenting with biliary tract manifestations simulating lymphoproliferative disease and adenocarcinoma, respectively. Clinical, radiological, and histopathological findings ultimately led to the correct diagnosis, and revealed useful nuances for detection of future cases. Application of specific criteria, such as the classic Japan Biliary Association clinical diagnostic criteria published in 2012 and revised in 2020 as well as the 2019 American College of Rheumatology and European League Against Rheumatism (ACR/EULAR) classification criteria, has limitations but provides important warnings to be considered in the diagnostic journey of these challenging cases.
Keywords: IgG4; Cholangitis; Cholangiocarcinoma; Pancreatitis
| Introduction | ▴Top |
Immunoglobulin G4-related disease (IgG4-RD) was first defined as a nosological entity in 2003, as an umbrella designation to encompass a range of complex, interlinked clinical conditions including autoimmune pancreatitis, retroperitoneal fibrosis, Mikulicz’s disease, pulmonary fibrosis, and lymphadenopathy with infiltrates rich in IgG4-positive plasma cells [1, 2].
The classic presentation of IgG4-RD involves the development of fibrosis and inflammatory mass lesions in the periampullary region as age advances, with a differential diagnosis strongly suggestive of malignancy. In the liver, parenchymal involvement mimicking autoimmune hepatitis has been described, but the most relevant and challenging aspect for the clinician is to distinguish IgG4-related hepatobiliary involvement from cholangiocarcinoma [1]. Although this presentation is rare, proper diagnosis is essential, as it can spare patients the high morbidity and mortality of surgical intervention and allow appropriate treatment, which consists of corticosteroid therapy [3, 4].
At the outpatient pancreas clinic of Hospital de Base do Distrito Federal (HBDF), Brasilia, Brazil, we have followed several cases of patients referred with a tentative diagnosis of cholangiocarcinoma, lymphoma, or periampullary cancer who were ultimately found to have IgG4-RD. Methods for the diagnostic elucidation of IgG4-RD must be disseminated more widely, to ensure that physicians maintain an appropriate level of diagnostic suspicion and include these diseases systematically in their differential diagnose, thus improving cure rates and minimizing the potential for iatrogenic injury from unnecessary procedures.
In this article, we report two cases in which heightened diagnostic suspicion allowed a more appropriate initial response, management, and follow-up of these patients. We also comment on the diagnostic process itself, based on the use of the Japan Biliary Association clinical diagnostic criteria [1] and the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria [2]. The ACR/EULAR criteria in particular were conceived to guide the inclusion of patients in more complex research designs, such as case-control studies and meta-analyses; however, as is common practice for the clinician, we will extrapolate them to individual cases. We observe the limitations of these criteria in clinical practice, as indicated by their authors, and we emphasize the importance of a rigorous method for a more assertive diagnosis.
This study was approved by the relevant institutional review board (Comite de Etica em Pesquisa do Instituto de Gestao Estrategica em Saude do Distrito Federal, IGESDF) with opinion number 06512819.0.00008153.
| Case Reports | ▴Top |
Case 1
Investigations
A 56-year-old woman presented with a history of chronic abdominal pain, especially in the mid-abdomen and left flank. She had been referred for a second opinion after a recent hospital admission for investigation of a wasting syndrome with hypertrophy of the submandibular glands, during which left axillary lymphadenopathy and multiple pulmonary nodules were identified. She had been evaluated by rheumatology and pulmonology, with diagnostic hypotheses including lymphoma, rheumatoid arthritis, Sjogren’s syndrome, and sarcoidosis. She reported no noteworthy events in her past or family pathological history.
Diagnosis
Lymph node biopsy was negative for malignancy. Immunohistochemistry for markers such as CD20/CD3/CD10 was performed, and the panel was negative for malignancy and compatible with reactive lymphoid hyperplasia with areas of thickening. The specimen was deemed inappropriate for a second immunohistochemistry for IgG4 due to inadequate storage of the paraffin block, and only nonspecific inflammation was found. Laboratory tests revealed an alkaline phosphatase (ALP) level of 400 U/L (normal if up to 120 U/L), gamma-glutamyl transferase (GGT) of 200 U/L (normal if up to 37 U/L), total bilirubin of 2 mg/dL (normal if up to 1.2 mg/dL), and IgG4 of 3,460 mg/dL (normal if up to 88 mg/dL). Hepatitis B and C serologies were negative, albumin was 4 g/dL, prothrombin time was 96% of normal, and international normalized ratio was 1.1. Then the patient was referred to gastrointestinal evaluation.
Given the clinical suspicion of lymphomas and IgG4-associated disease, a positron emission tomography-computed tomography (PET-CT) scan was performed, showing fluorodeoxyglucose (FDG)-avid lesions in several lymph node chains and the pulmonary and hepatic parenchyma, but most significantly in the submandibular glands, as well as pancreatic enlargement with increased FDG uptake (Fig. 1). Considering marked biliary tree involvement seen on magnetic resonance imaging (MRI) (Fig. 2) and elevated IgG4 level, a diagnosis of IgG4-RD with autoimmune pancreatitis and IgG4-associated cholangitis was made based on the Japan Biliary Association criteria (biliary imaging finding, IgG4, and association with autoimmune pancreatitis) and on the international consensus diagnostic criteria (ICDC) for autoimmune pancreatitis (imaging, serum IgG4, association with submandibular gland enlargement on physical examination, and imaging evidence compatible with IgG4-associated cholangitis) [1, 3].
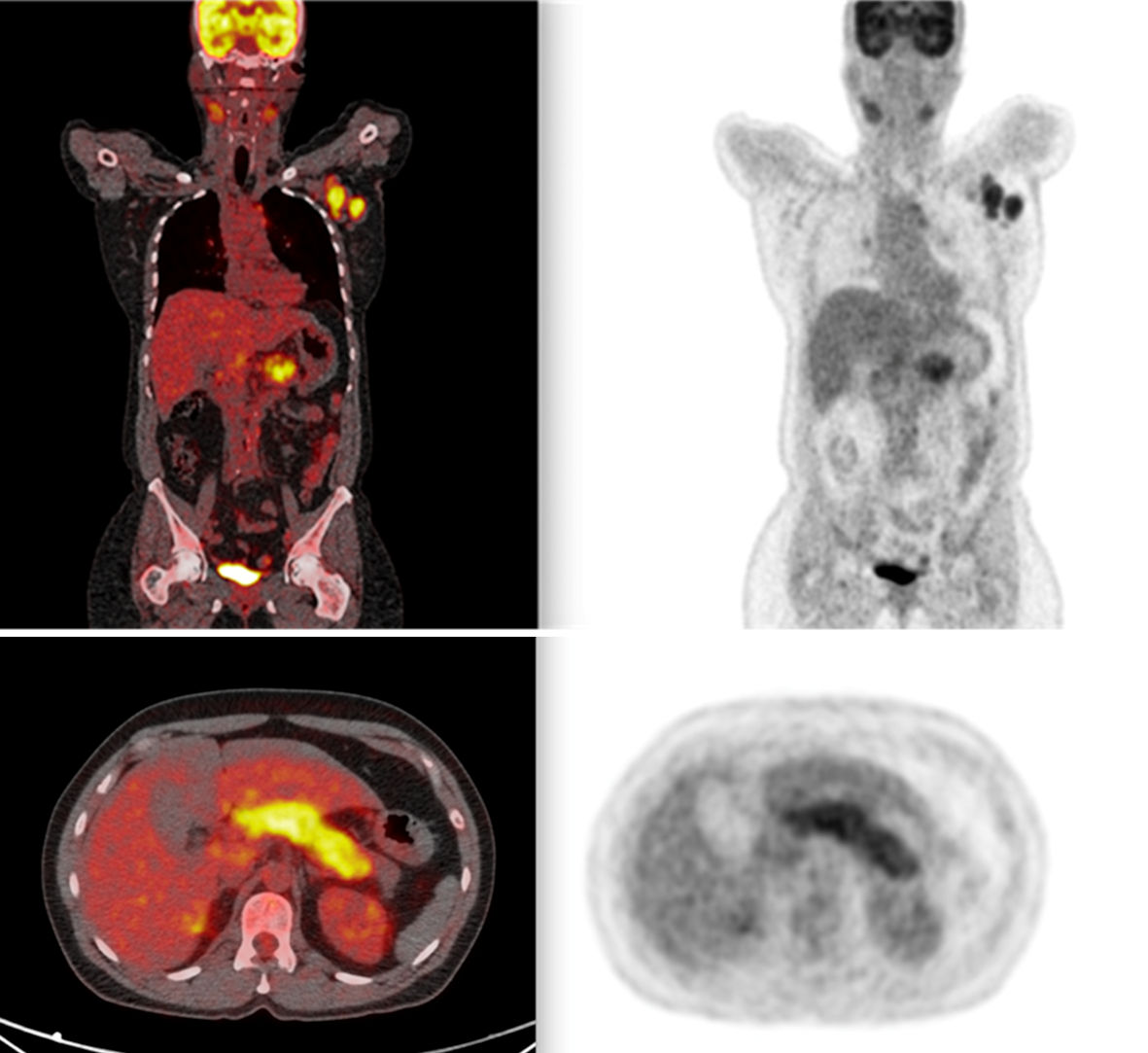 Click for large image | Figure 1. PET scan showing widespread lymph node involvement (pancreatic, submandibular, and hepatic hilum). In the pancreas, in addition to pancreatic enlargement, note the increased FDG uptake. PET: positron emission tomography; FDG: fluorodeoxyglucose. |
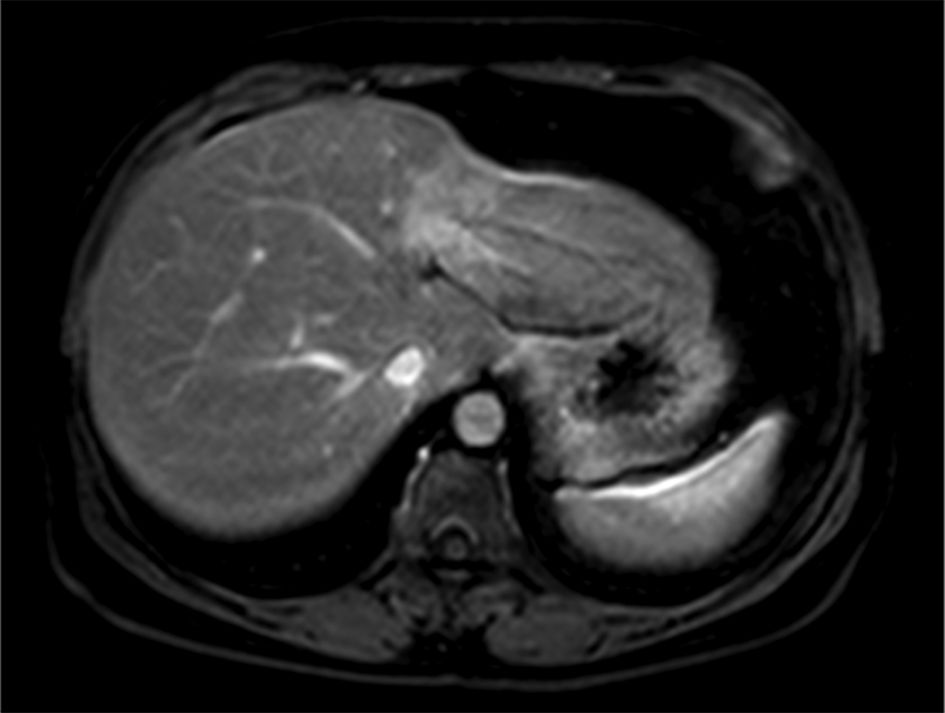 Click for large image | Figure 2. Narrowing of the left biliary tree with retraction of the hepatic parenchyma. |
Treatment
Prednisone 40 mg was initiated, with significant improvement of the lymphadenopathy and pulmonary lesions and complete resolution of biliary involvement on MRI.
Follow-up and outcomes
She is being followed jointly by the rheumatology, pulmonology, and pancreas clinics, and continues to take 10 mg prednisone daily. This patient’s ACR/EULAR score was 42 (IgG4 > 5 times the upper limit of normal, parotid involvement, and pancreatic and biliary involvement), and she did not meet any of the exclusion criteria, notably malignancy, infection, and positive screening for other rheumatologic diseases or presence of inflammatory bowel disease (normal colonoscopy). The test results shown in Table 1 referring to admission and latest outpatient visit demonstrate an improvement in cholestasis markers, but GGT remains increased due to hepatic steatosis that the patient developed.
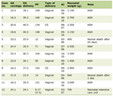 Click to view | Table 1. Laboratory Findings of Case 1 |
Case 2
Investigations
A 65-year-old man was referred with a history of early satiety, postprandial fullness, anorexia, and weight loss (approximately 25 kg in 4 months). He later developed palmoplantar pruritus, jaundice, choluria, and acholia as well as an unintended weight loss of approximately 7 kg during that month alone. The patient denied any alcohol or tobacco use. On physical examination, he was emaciated, weighing approximately 50 kg, and jaundiced without palpable masses in the abdomen or lymph node enlargement. He reported no noteworthy events in his past or family pathological history.
Diagnosis
A CT scan of the abdomen revealed a solitary hypovascular nodule at the confluence of the common hepatic duct. On admission, the total bilirubin level was 10 mg/dL (direct bilirubin 6 mg/dL) (normal if up to 1.2 mg/dL and 0.8 mg/dL, respectively) ALP 1,080 U/L (normal if up to 120 U/L), and GGT 104 U/L (normal if up to 37 U/L). A diagnostic hypothesis of cholangiocarcinoma was raised.
An open biopsy of the liver and celiac lymph nodes was performed. The mass was deemed unresectable; the decision was thus made to obtain specimens of a segment V hepatic nodule and celiac lymph node for frozen-section analysis. Histopathological examination of the biopsy specimens was consistent with cholestasis and reactive lymphadenopathy. There was no evidence of malignancy.
Endoscopic retrograde cholangiopancreatography (ERCP) was performed with the use of fluoroscopy through an external biliary drain, which had been placed in an attempt to decompress the biliary tree 1 month earlier, for placement of a self-expandable metallic stent. Papillotomy and removal of microcalculi and biliary sludge were necessary to complete the procedure (Fig. 3).
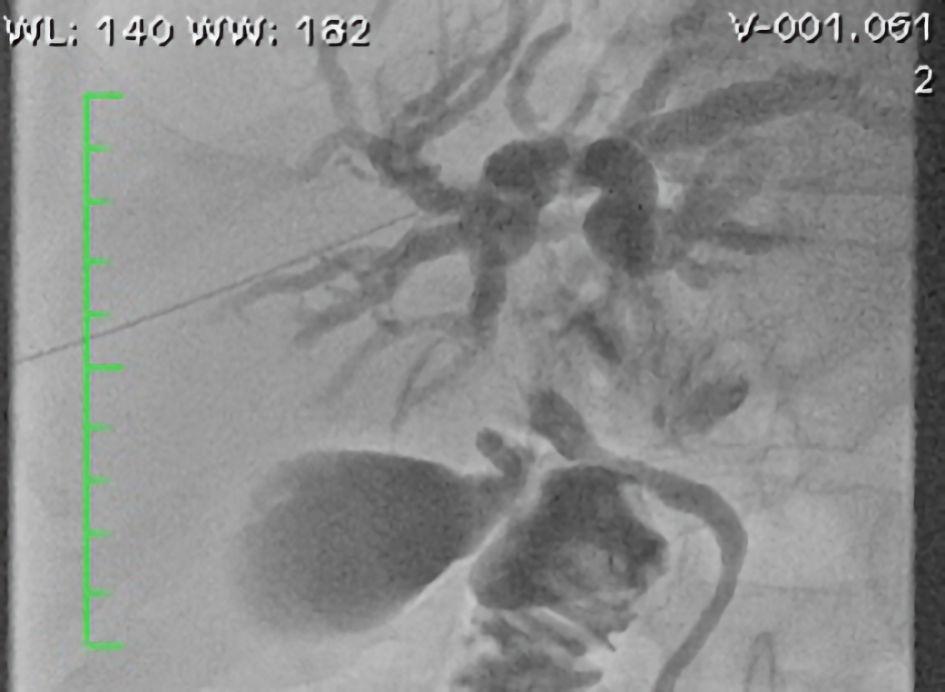 Click for large image | Figure 3. Fluoroscopy showing compression at the confluence of the hepatic ducts. |
After discharge, during an outpatient appointment, the diagnostic hypothesis of IgG4-associated cholangitis was first considered. The patient’s IgG4 level was 2,290 mg/dL, and a review of imaging tests (CT and MRI) showed an enlarged pancreas with narrowing of the duct of Wirsung suggestive of autoimmune pancreatitis. A reassessment of the liver biopsy findings was requested, which revealed hepatic parenchyma with a mixed inflammatory infiltrate of lymphocytes, plasma cells, neutrophils, and eosinophils detected mainly in the portal space, sometimes with lymphoid accumulations. The inflammatory infiltrate was invading the bile ducts, with a wide spectrum ranging from portal spaces with well-preserved ducts to distorted ducts with discontinuity of the basement membrane, occasionally with disrupted epithelial cells. Fibrotic areas in a storiform pattern of fibrosis were also observed, with no remaining structures. Foci of inflammatory infiltrates were observed in terminal venules. The liver lobes showed cholestasis and occasional lymphocytes in the sinusoids. Immunohistochemistry was as previously reported, with > 10 IgG4+ plasma cells per high-power field, reaching the conclusion of IgG4-associated sclerosing cholangitis and reactive follicular lymphoid hyperplasia in addition to storiform fibrosis (Fig. 4). The patient met histological criteria for IgG4-associated cholangitis according to the Japanese criteria and for autoimmune pancreatitis according to the ICDC (enlarged pancreas, association with histopathological findings including immunohistochemistry for IgG4 in extrapancreatic tissues, and serum IgG4).
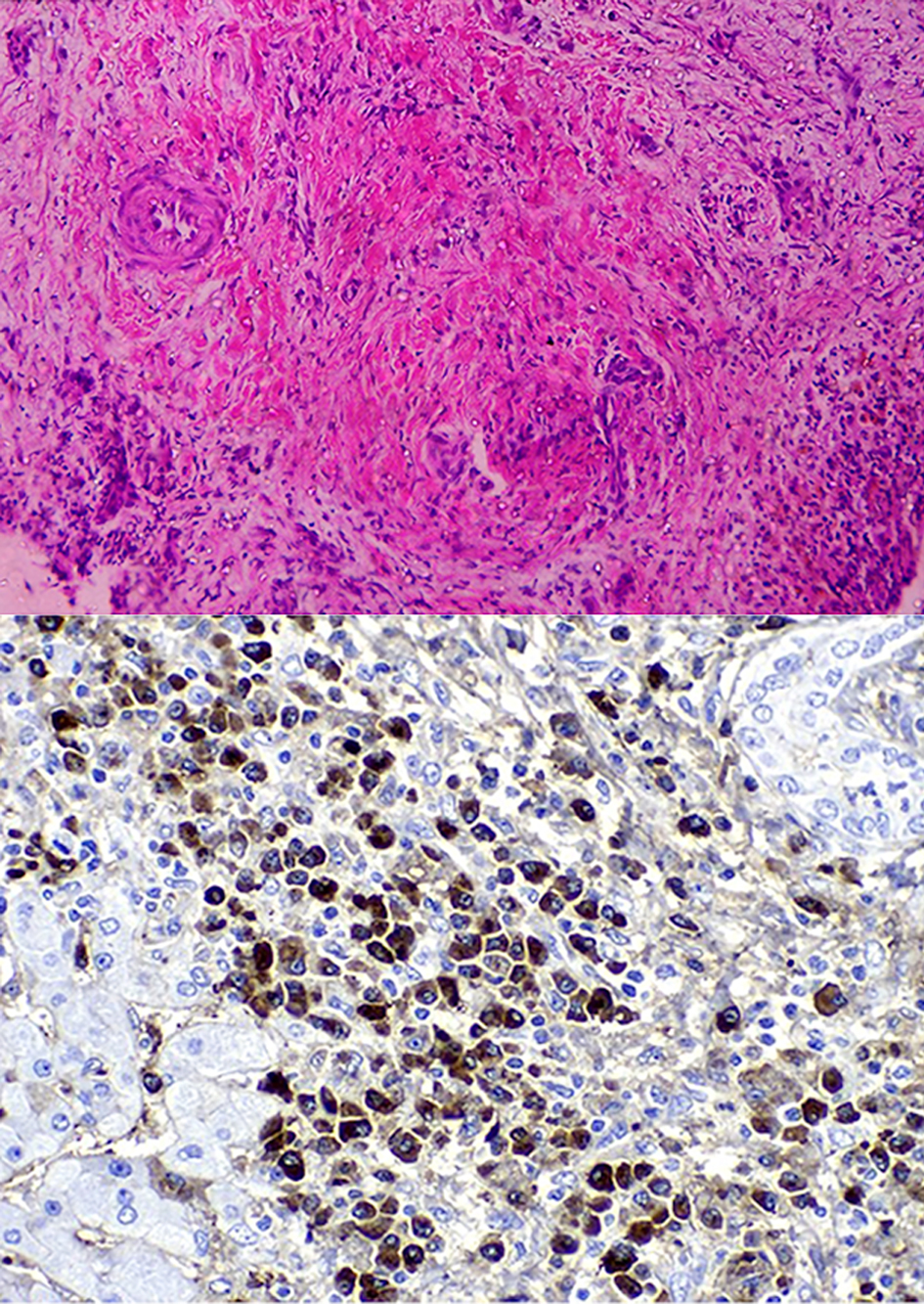 Click for large image | Figure 4. Review of original biopsy, with evidence of storiform fibrosis on hematoxylin and eosin staining and IgG4+ plasma cells on immunohistochemistry. Ig: immunoglobulin. |
Treatment
A single dose of ivermectin (two 6-mg tablets) was administered to prevent hyperinfection in the setting of high-dose corticosteroid therapy, and the patient was started on prednisolone 40 mg daily for 1 month. Within 2 weeks, he was asymptomatic, and was tapered off prednisolone due to marked dysglycemia (fasting blood glucose 346 mg/dL).
Follow-up and outcomes
MR cholangiography showed slight dilation of the bile ducts and evidence of stent obstruction. Laboratory tests showed increased liver and biliary enzymes and a fasting blood glucose of 275 mg/dL. Metformin 500 mg twice daily was started. Ursodeoxycholic acid (300 mg once daily) was added to the patient’s drug regimen (prednisone 30 mg once a day (QD), metformin 500 mg twice a day (BID), gliclazide 60 mg QD in the morning). Although emaciated (current weight 46.5 kg), the patient had no complaints and reported restoration of normal sleep and appetite patterns. He was readmitted to undergo further surgery for removal of the self-expandable metallic stent and biliointestinal bypass.
The test results shown in Table 2 referring to admission and latest outpatient visit demonstrate an improvement in bilirubin levels, especially after removal of the prosthesis. However, cholestasis markers remain increased, which shows that small-duct disease persists.
Click to view | Table 2. Laboratory Findings of Case 2 |
This patient’s ACR/EULAR score was 44 (IgG4 > 5 times upper limit of normal; 50% IgG4+/IgG+ ratio on immunostaining; up to 35 IgG4+ plasma cells per high-power field; storiform fibrosis; pancreatic and biliary tree involvement). The exclusion criterion of fundamental clinical interest was to rule out malignancy.
| Discussion | ▴Top |
IgG4 is the smallest fraction of IgG. Whether it is actually pathogenic remains a matter of controversy, given its lability and difficulty in activating complement; however, it is recognized as an important marker of inflammatory processes, especially those related to hypersensitivity and fibrosis, for reasons which have yet to be fully elucidated [3, 4].
In the biliary tree, IgG4-RD may present as one of two phenotypes: inflammatory (simulating a mass lesion with stricture) and stenosing. The clinical picture is that of extrahepatic cholestasis, with signs and symptoms such as pruritus, jaundice, pain, infection, and atrophy of the affected lobe of the liver. Comorbidity with other autoimmune diseases (especially autoimmune pancreatitis, retroperitoneal fibrosis, and Mikulicz’s syndrome) and with hypergammaglobulinemia (especially of the IgG4 fraction) are important diagnostic clues. One pitfall is that the same findings are often present in patients with biliary tract cancers, effectively simulating the Bismuth classification for cholangiocarcinoma (a condition with a very narrow diagnostic window for curative therapy) [5, 6]. Thus, rigorous international criteria were established, with advice for teams to improve their expertise in differential diagnosis or risk undertreating the rapidly lethal disease that is cholangiocarcinoma [5]. Both the Japanese and US/European criteria exclude a diagnosis of IgG4-RD if malignancy has not been adequately ruled out.
IgG4-RD is characterized by a multiorgan fibroinflammatory disorder, with cholangitis being its major biliary manifestation. Consequent to hepatic involvement, the biliary tree is affected by the key features of IgG4 disease: lymphoplasmacytic infiltrates, a characteristic storiform pattern of fibrosis, and obliterative phlebitis, which may cause secondary sclerosing cholangitis (SSC) [5]. Quantification of serum IgG4 can be of diagnostic utility (levels are elevated in 60% of patients), as can detection of an IgG4+/IgG+ cell ratio > 40% in tissues. Obstructive jaundice is one of the most common manifestations [4], and clinical examination is often consistent with pseudotumor [5]. Corticosteroids are the mainstay of treatment, and response is associated with good prognosis. However, discontinuation or even tapering leads to relapse in approximately 30% of cases [1, 5].
The two cases described herein shared a common feature, pancreatic involvement. Both patients met ICDC and HISORt criteria and ICDC [3, 6, 7] for autoimmune pancreatitis, but at least in case 2, the differential diagnosis of IgG4-RD versus cholangiocarcinoma was especially challenging.
As noted above, the presentation of hepatobiliary IgG4-RD may be predominantly stenotic/fibrotic or inflammatory. In case 2, fibrosis was predominant, ultimately requiring continuous biliary drainage with a self-expandable metallic stent. In a series of 54 patients already on corticosteroid treatment, biliary endoprosthesis placement was safe and effective [8]. In our case, however, the initiation of steroid therapy was delayed, and the stenosis required surgical intervention, due to progressive disease with demonstrable invasion of the stent by fibrous tissue. This patient still has small-duct disease, persistently high levels of ALP and GGT, and is dependent on ursodeoxycholic acid and prednisone (10 mg daily), but 3 years after diagnosis and 1 year after surgery, he remains free of biliary cirrhosis.
In case 1, the multitude of affected organ systems was very helpful in establishing the proper diagnosis. Nevertheless, the patient’s experience still involved a litany of specialists, including rheumatologists and pulmonologists, and multiple tentative diagnoses, including Sjogren’s syndrome and sarcoidosis [9]. Ultimately, the unique distribution of involvement (salivary glands, lymph nodes, pancreas, and biliary tract), the use of the 2012 Japan Biliary Association criteria and PET-CT findings were instrumental in clinching the diagnosis of IgG4-RD [9, 10]. Combining the Japanese and ACR/EULAR criteria would have diagnosed the present cases quite assertively.
Still on the topic of differential diagnosis, our two cases underwent colonoscopy without abnormal findings that could suggest ulcerative colitis and thus raise the suspicion of primary sclerosing cholangitis. This step in the diagnostic journey has been observed by the Japan Biliary Association and by the ACR/EULAR, although it is not mandatory or excluding for routine diagnosis [1-4].
Despite the European and American Society of Rheumatology recommendation that diagnosis be considered on a case-by-case basis, given the high specificity of the ACR/EULAR criteria and their proven utility for screening of large groups for inclusion in complex study designs such as meta-analyses, we believe these criteria can be used in clinical practice for diagnosis of individual patients. In the cases we described, these criteria were very useful, especially in determining that ruling out malignancy should be the first concern.
Conclusions
IgG4-RD is a recently described disease entity and a particular diagnostic challenge, especially because of its ability to mimic malignant neoplasms and inflammatory diseases that are more commonly managed by different specialties. As biliary involvement is common in the IgG4-RD spectrum, a high level of clinical suspicion and thorough knowledge of diagnostic criteria are essential for proper management and avoidance of iatrogenic events. Ruling out malignancy and ensuring correct diagnosis of autoimmune pancreatitis are crucial. With some restrictions, we believe that the ACR/EULAR criteria can also be useful in daily clinical practice, but the Japan Biliary Association criteria remain the most useful and recommended in clinical practice for patients with biliary involvement.
Acknowledgments
None to declare.
Financial Disclosure
The authors have no financial relationships relevant to this article to disclose.
Conflict of Interest
The authors have no conflicts of interest to disclose.
Informed Consent
Informed consents were obtained from the two participants included in the study.
Author Contributions
Conception and design: Ricardo Jacaranda de Faria and Marcos Vasconcelos Carneiro. Analysis and interpretation: Ricardo Jacaranda de Faria and Marcos Vasconcelos Carneiro. Data collection: Raissa Dytz, Gabriela dos Santos, and Lucio Lucas Pereira. Writing the article and critical revision of the article: all authors. Overall responsibility: Ricardo Jacaranda de Faria. All authors have read and approved the final version of the article.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
ACR/EULAR: American College of Rheumatology and European League Against Rheumatism; ALP: alkaline phosphatase; IgG4-RD: immunoglobulin G4-related disease; SSC: secondary sclerosing cholangitis
| References | ▴Top |
- Nakazawa T, Kamisawa T, Okazaki K, Kawa S, Tazuma S, Nishino T, Inoue D, et al. Clinical diagnostic criteria for IgG4-related sclerosing cholangitis 2020: (Revision of the clinical diagnostic criteria for IgG4-related sclerosing cholangitis 2012). J Hepatobiliary Pancreat Sci. 2021;28(3):235-242.
doi pubmed - Wallace ZS, Naden RP, Chari S, Choi HK, Della-Torre E, Dicaire JF, Hart PA, et al. The 2019 American College of Rheumatology/European League against rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis. 2020;79(1):77-87.
doi pubmed - Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, Kim MH, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40(3):352-358.
doi pubmed - Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539-551.
doi pubmed - Martinez-Valle F, Riveiro-Barciela M, Salcedo MT, Merino-Casabiel X, Fernandez-Codina A, Torres I, Esteban R, et al. Sclerosing cholangitis related to IgG4: not always a curable entity. Ann Hepatol. 2019;18(1):215-219.
doi pubmed - Roos E, Hubers LM, Coelen RJS, Doorenspleet ME, de Vries N, Verheij J, Beuers U, et al. IgG4-associated cholangitis in patients resected for presumed perihilar cholangiocarcinoma: a 30-year tertiary care experience. Am J Gastroenterol. 2018;113(5):765-772.
doi pubmed - Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing the Mayo Clinic's HISORt criteria. J Gastroenterol. 2007;42(Suppl 18):39-41.
doi pubmed - Kuraishi Y, Muraki T, Ashihara N, Ozawa M, Nakamura A, Watanabe T, Ito T, et al. Validity and safety of endoscopic biliary stenting for biliary stricture associated with IgG4-related pancreatobiliary disease during steroid therapy. Endosc Int Open. 2019;7(11):E1410-E1418.
doi pubmed - Ozawa Y, Yamamoto H, Yasuo M, Komatsu M, Ushiki A, Hamano H, Uehara T, et al. A comparison of the features of fluorine-18 fluorodeoxyglucose-positron emission tomography (FDG-PET) between IgG4-related disease with bilateral hilar lymphadenopathy and sarcoidosis. Nagoya J Med Sci. 2020;82(1):101-111.
- Okuda H, Yamamoto Y, Mitamura K, Norikane T, Nishiyama Y. F-18 FDG PET/CT to monitor disease in patients with IgG4-related disease before and after therapy. J Nucl Med. 2018;58(supp 1):592.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.