

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 11, Number 12, December 2020, pages 388-393
Local Lung Mass Masquerading a Very Aggressive Extraskeletal Ewing Sarcoma Presenting as Bilateral Paraparesis in a Young Adult
Mytri Pokala, e, Vikram Sangania, Mamtha Ballab, Ganesh Prasad Meruguc, Venu Madhav Konalad
aDepartment of Internal Medicine, Quantum HC, Navicent Health, Macon, GA 31201, USA
bDepartment of Internal Medicine, University of Toledo and Promedica Toledo Hospital, Toledo, OH 43606, USA
cDivision of Geriatric Medicine, Department of Family Medicine, University of Toledo, Toledo, OH 43614, USA
dDivision of Medical Oncology, Department of Internal Medicine, Ashland Bellefonte Cancer Center, 122 St Christopher Dr, Ashland, KY 41169, USA
eCorresponding Author: Mytri Pokal, Department of Internal Medicine, Quantum HC, Navicent Health, 777 Hemlock street, Macon, GA 31201, USA
Manuscript submitted September 9, 2020, accepted September 23, 2020, published online October 21, 2020
Short title: An Extraskeletal Ewing Sarcoma
doi: https://doi.org/10.14740/jmc3582
| Abstract | ▴Top |
Ewing sarcoma is typically seen in children involving long bones. Although well described, its presentation in extraskeletal tissues is relatively rare and is classified as an Ewing sarcoma family of tumors. They are mostly curable when they occur in children. An extraskeletal Ewing sarcoma in adults is uncommon, limiting the experience in adult oncologists. The biopsy is essential for definitive diagnosis, which shows small round blue cells that must be differentiated from lymphoma, embryonal rhabdomyosarcoma, and small cell carcinoma. Management is multimodal, involving surgery, radiation for local treatment of primary tumor, and systemic chemotherapy. A multidisciplinary approach, coupled with risk-adapted intensive neoadjuvant and adjuvant multi-agent chemotherapies and other modalities such as radiation and surgery for control of the primary site and metastatic disease, is needed. The primary multidrug chemotherapy regimen consists of alternating cycles of vincristine/doxorubicin/cyclophosphamide (VDC) and ifosfamide/etoposide (IE) given every 2 weeks with growth factor support. Prognosis and the 5-year survival rate are better for localized than the metastatic disease, and in metastatic disease, it is better for patients with lung metastasis than other metastatic disease sites. We describe a rare extraskeletal tumor arising from a lung that tested positive for Ewing sarcoma, also known as Askin’s tumor in a young adult. In our case, the tumor rapidly metastasized locally to involve the thoracic spine causing paraparesis. Timely diagnosis and early management are essential to improve outcomes. We also present how treatment can be delayed due to sepsis and emphasize the careful multispecialty approach’s importance.
Keywords: Extraskeletal Ewing sarcoma; PNET; Askin’s tumor; Thoracic spine metastasis
| Introduction | ▴Top |
Ewing sarcoma is a very aggressive bone tumor primarily seen in adolescence and peaks in the second decade of life. Atypically primitive neuroectodermal tumors (PNETs) involving various soft tissues like the chest wall, lungs, visceral pleura, oral cavity, and perineum have been described, which together with Ewing sarcoma, are classified as Ewing’s family of tumors. It is mostly curable in children. However, the prognosis is poor with increasing age [1]. We describe a rare case of extraskeletal PNET initially presented as a local lung tumor that rapidly involved the adjacent spine, causing paraparesis in a young adult. We also described how sepsis posed a challenge in treating this locally aggressive tumor, making prognosis grim.
| Case Report | ▴Top |
A 27-year-old gentleman with no significant past medical history presented with hoarseness of voice for 1 month. He was diagnosed with laryngitis and was prescribed antibiotics. His symptoms continued to worsen with cough and upper back pain, and decided to come to the emergency room. He has not had a significant cough but had an episode of “tar-like” mucus once associated with nasal congestion and hoarseness. He denies fever, chills, hemoptysis, body aches, nausea, vomiting, or other symptoms. He denies sick contacts or recent travel. He had no prior surgeries. His family history was significant for breast cancer in grandmother and colon cancer in grandfather. He smoked cigarettes on and off for the last 10 years and quit 2 weeks ago. He denies alcohol use but admits to occasional marijuana use.
On admission, his vitals are temperature 37.4 °C oral, heart rate of 108 beats per min, respiratory rate of 20 breaths per min, blood pressure of 146/101 mm Hg, and oxygen saturation 100% on 2 L via nasal cannula. On examination, he is an alert, well-built young gentleman with a body mass index (BMI) of 24. On auscultation of the chest had decreased breath sounds at the left base. Otherwise, the rest of the exam is unremarkable. Admission labs are summarized in Table 1. Chest X-ray showed left lower lobe effusion vs. mass.
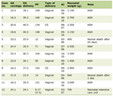 Click to view | Table 1. Admission Labs |
Hospital course
Based on tachycardia, elevated white blood cell count in the emergency room (ER), and abnormal chest finding with a presentation of cough and shortness of breath, a presumed diagnosis of pneumonia was made in ER with no response to outpatient antibiotic treatment. He was started on broad-spectrum antibiotic vancomycin and piperacillin/tazobactam, and blood cultures obtained. He was admitted for further management. Computed tomography (CT) of the thorax was done that revealed large multilocular cystic and solid left upper lobe mass concerning neoplasm with left to right mediastinal shift and compressive atelectasis (Fig. 1). There is also evidence of large pleural effusion on the left side with ongoing respiratory compromise and concern for hydropneumothorax. A chest tube was placed, fluid from the Fuhrman catheter sent for analysis. Based on light’s criteria, fluid was determined to be exudative. Although cytology was negative for malignant cells, cardiothoracic surgery was consulted. They performed a thoracoscopic examination, finding pleural studding in addition to the mass had undergone Left video-assisted thoracoscopic surgery and pleural biopsy with preliminary pathology confirming poorly differentiated malignant neoplasm with the final pathology pending. He was presumed to have local lung cancer. The chest tube was changed to Pleurx. It was recommended to follow up outpatient with a positron emission tomography (PET) scan scheduled. He completed an antibiotic course. He also developed left upper extremity swelling during hospitalization, confirming cephalic vein thrombosis, and was discharged on low dose Eliquis.
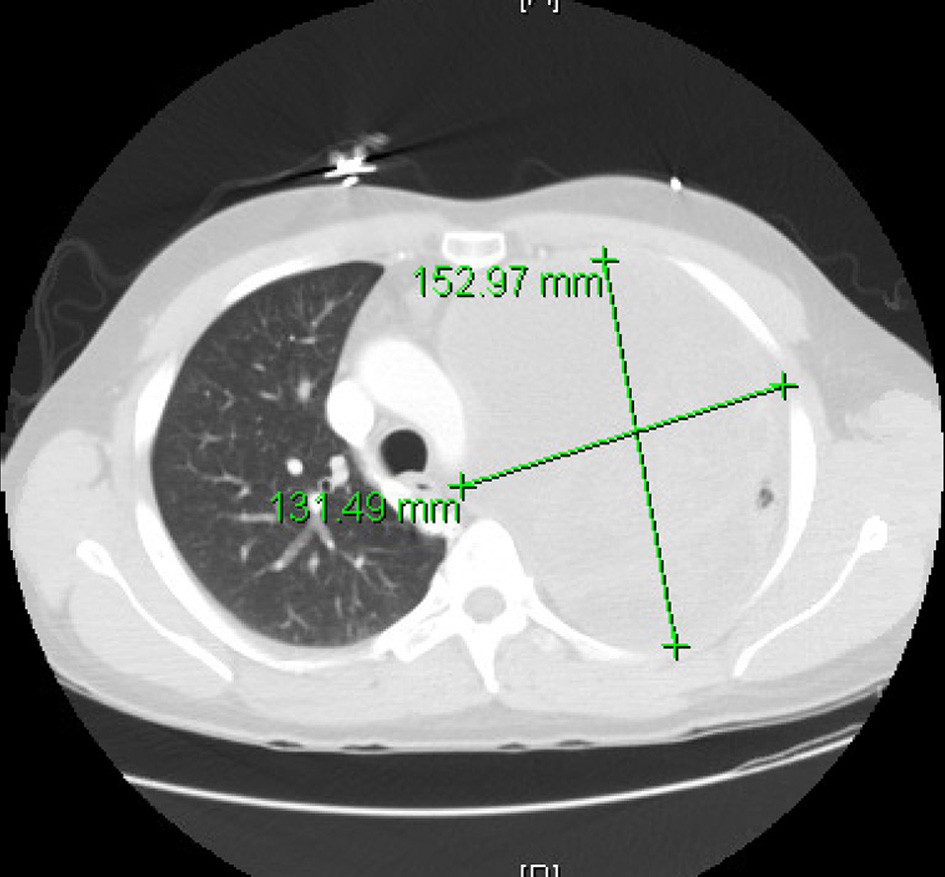 Click for large image | Figure 1. Computed tomography (CT) thorax showing left lung mass with a left to right mediastinal shift. |
The outpatient PET scan showed hypermetabolic uptake of left chest mass with several nodular hypermetabolic foci in the left pleural space, likely representing regional pleural-based neoplasia, no apparent hypermetabolic disease outside the confines of the chest.
Ten days later, he comes back to the ER with a weakness of the right lower extremity associated with decreased mobility. The patient reports that he has lost the ability to plantar/dorsiflexion in his right lower extremity. His sensation was intact and denied incontinence of bowel or bladder. Magnetic resonance imaging (MRI) of the thoracic and lumbar spine was done, which revealed metastatic lesion to the thoracic area with substantial spinal cord compromise (Fig. 2). The patient was started on dexamethasone. Neurosurgery was consulted, and he underwent posterior decompression and removal of the tumor from thoracic vertebrae 2 - 3 with partial T1 - T4 decompression.
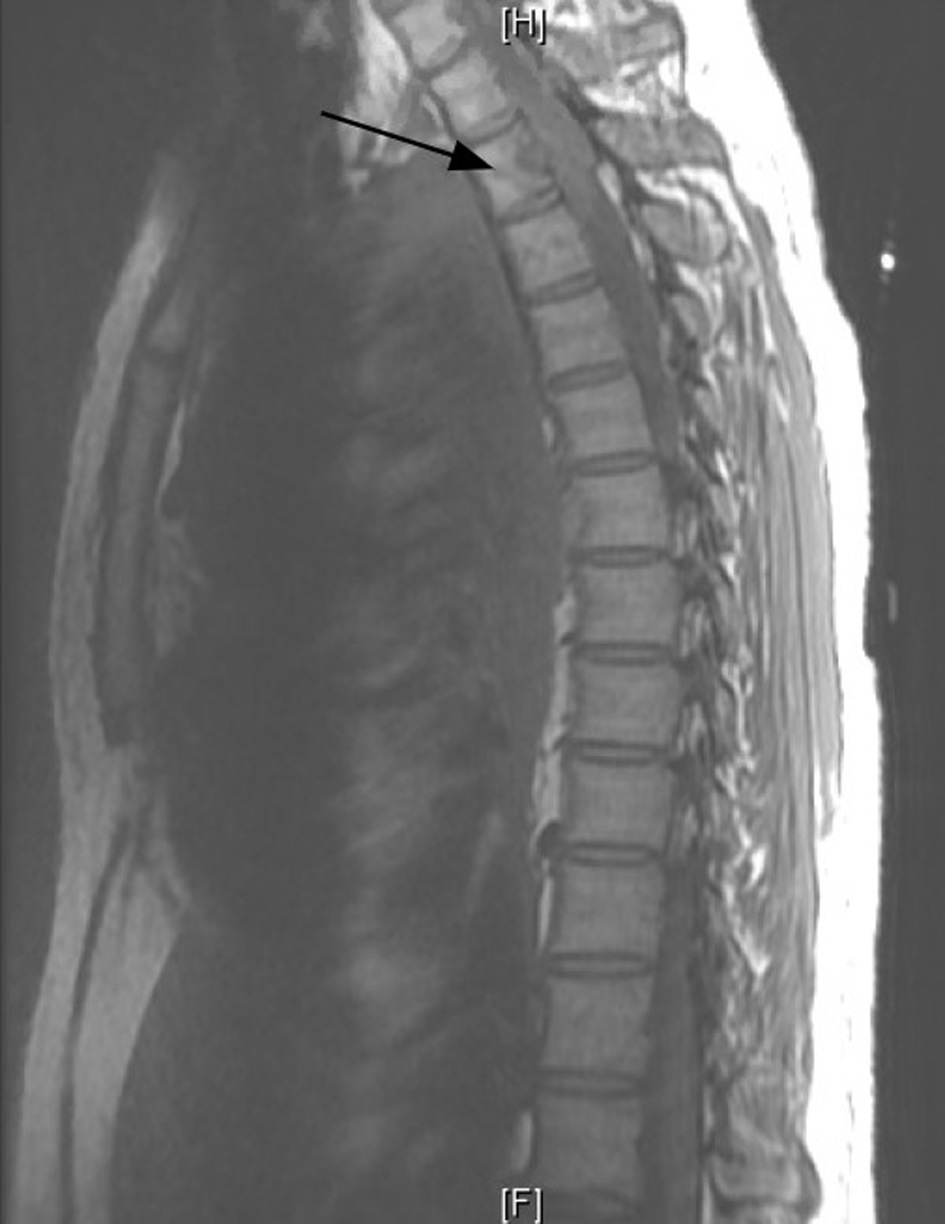 Click for large image | Figure 2. Magnetic resonance imaging (MRI) thoracic spine without contrast. Infiltration of the T2 vertebral body and pedicle by the known malignancy in the left lung and pleural space (arrow). |
Pathology from the left pleural implant from the previous admission showed a small round blue cell tumor compatible with Ewing sarcoma (Fig. 3). Immunohistochemically stained slides show that the tumor is positive for pan-keratin. Vimentin, synaptophysin, cluster of differentiation 99 (CD99), friend leukemia integration 1 transcription factor (FLI1), AE1/AE3 (anti-cytokeratin antibody, scattered cells) and negative for CD45, smooth muscle actin (SMA), CD20, cytokeratin 7 (CK7), CK20, SRY-related HMG-box 10 (SOX10), P40 and thyroid transcription factor 1 (TTF1). Fluorescence in situ hybridization (FISH) is positive for Ewing sarcoma breakpoint region 1 (EWSR1) gene rearrangement in 78% of cells involving the chromosomal translocation of the EWSR1 gene on chromosome 22q12.
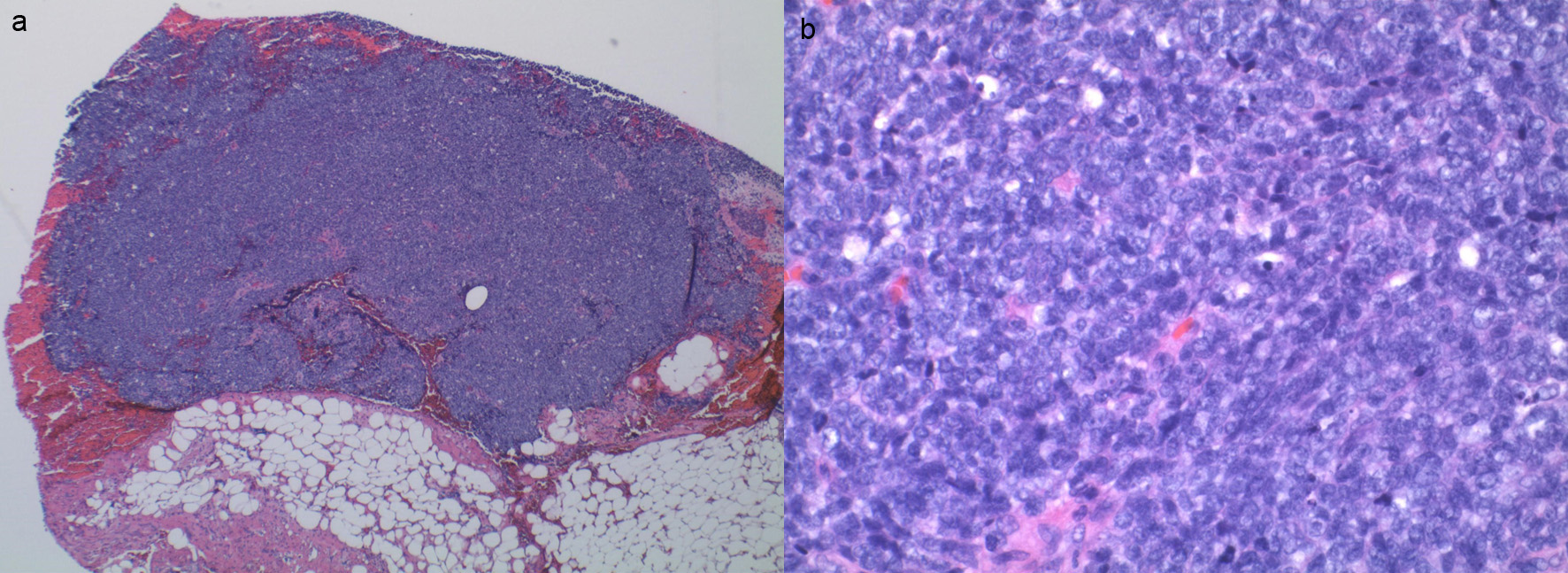 Click for large image | Figure 3. (a) Sheets of small round blue cell tumor without necrosis. (b) Small round blue cells. |
Flow cytometry studies after erythroid lysis revealed a B-cell population consisting of predominantly small cells showing no clonality or antigenic aberrancy. The T cells show normal expression of the pan T-cell antigens and have a normal CD4: CD8 ratio. The natural killer (NK) cells account for 4.8 % of the total lymphocytes. A CD45-negative non-hematopoietic cell population is detected. Thoracic tumor biopsy again revealed small round blue cells, the FISH study eventually reported as tumor consistent with Ewing sarcoma.
Discussion in multidisciplinary tumor board radiation oncologists recommends starting palliative radiation to T1 - T3 as soon as possible to 3,000 cGy in 10 fractions while inpatient. Since Ewing sarcoma is primarily treated in the pediatric population, a pediatric oncologist was consulted. He then underwent bilateral bone marrow biopsy for further staging. Bilateral bone marrow aspirate smear, clot section, and core biopsy revealed normocellular marrow (40%) with trilineage hematopoiesis and no morphological evidence of involvement Ewing sarcoma. Flow cytometry showed no immunophenotypic evidence of B-cell or T-cell lymphoproliferative disorder, and no monoclonal plasma cell population identified, confirming no evidence of distant metastatic disease. Due to its locally aggressive nature, a multimodal approach was recommended that involved chemotherapy, local control with surgery, and radiation. Urology was consulted for fertility preservation and echocardiogram to document baseline cardiac function was done before chemotherapy.
The patient was discussed in the tumor board. The plan was to administer multidrug chemotherapy with vincristine, doxorubicin, and cyclophosphamide (VDC) for cycle 1 followed by about six cycles of VDC, ifosfamide, and etoposide (VDC/IE) every 2 weeks followed by local control to include surgery along with radiation to hemithorax and spine. This will be followed by consolidative chemotherapy with eight more cycles of VDC/IE. Repeat imaging of the tumor to be planned after cycle 5 or 6 so that surgery/radiation can be planned accordingly. Neurosurgery recommended physical therapy with a thoracic-lumbar-sacral orthosis (TLSO) brace.
After the first chemotherapy, the patient started to develop sepsis-like syndrome. Infectious disease was consulted, and the patient was started empirically on antibiotics. Blood cultures were positive for staphylococcus with source likely thought to be post-operative wound infection. Chemotherapy was held, and infection was aggressively managed by local incision and drainage, wound vac placement followed by wound care, and prolonged intravenous vancomycin and Bactrim for Pneumocystis pneumonia (PCP) prophylaxis. The patient was able to complete cycle 1 and part A of cycle 2 and was discharged home to return for cycle 2, part B (IE).
Chemotherapy, radiation, or any surgical management were held because of sepsis, which is concerning, especially in this aggressive local tumor. A careful multisystem approach is necessary to improve outcomes that otherwise can make prognosis poor.
| Discussion | ▴Top |
Ewing sarcoma family of tumors includes classic Ewing sarcoma, PNET, extraosseous Ewing sarcoma, and Askin’s tumors (chest wall) have been described in the 1900s. They are aggressive sarcoma of bone and soft tissue. Classically they arise in long bones with a characteristic “onion peel” appearance on plain radiographs. It may occur in other bones and extraskeletal soft tissues. Ewing sarcoma tumors are a rare entity that is more common between 10 and 15 years of age, with an average annual incidence of 2.9 per million children younger than 20 years of age, slightly higher in males compared to females with a 6-fold higher incidence in white children than black children [2].
Reciprocal translocation of FLI1 gene on the long arm of chromosome 11 and EWSR1 on chromosome 22 is the primary cytogenetic abnormality believed to be involved in tumorigenesis. The biopsy is essential for definitive diagnosis, which shows small round blue cells that must be differentiated from lymphoma, embryonal rhabdomyosarcoma, and small cell carcinoma.
Periodic acid-Schiff (PAS) stain shows glycogen-filled cytoplasm. Immunohistochemical marker CD99 is sensitive, FLI1 fusion, and NK2 homeobox 2 (NKX2-2) are relatively sensitive, and specific to Ewing sarcoma.
Management is multimodal, involving surgery and radiation for local treatment of primary tumor and systemic chemotherapy. Chemotherapy is the mainstay often used in multiple cycles before and after surgery and radiation. A multidisciplinary team approach involving a combination of surgery, radiation, and multidrug chemotherapy have improved the 5-year survival rates to 60-73% in patients with localized disease [3] and 37-60% in patients with metastatic disease compared to 10% with surgery or radiation alone before the advent of multi-agent systemic therapy as per the literature review by Valdes et al. [4]. The 5-year overall survival for patients with isolated pulmonary metastatic disease is higher than patients with other sites of metastatic diseases, approximately 50% and less than 30%, respectively. Once the disease recurs, the patients tend to have a dismal prognosis [5].
In a retrospective review done by Valdes et al [4] involving 18 patients older than 19 years of age who received systemic therapy, the median overall survival for the entire group was approximately 21 months. The overall survival for patients with localized disease at 1, 2, and 3 years were 90%, 80%, and 70%, respectively. Their 1-year survival for patients with metastatic disease was 40%. These patients have Ewing sarcoma family of tumors with known extremity primaries with predominant soft tissue involvement [4]. In another retrospective review by Argon et al. involving 25 patients who are over 15 years, 20% of them have metastatic disease at presentation. The reported 2-year overall survival was 32.7% with poor prognosis in patients with localized pelvic primaries [6].
One of the strongest prognostic factors is a patient presenting with metastatic disease at diagnosis [5]. Several prognostic factors determine the intensity of therapy, including age, location of the primary tumor, localized or metastatic disease, chromosomal translocations, and response to therapy. For patients presenting with localized disease, primary tumors arising from the axial skeleton tend to have a worse prognosis than those arising from the extremity. In a series by Cotterill et al, the 5-year relapse-free survival was 61% and 40% with primary tumors arising from extremity and axial skeleton, respectively [7]. The induction therapy responses, as well as complete surgical resection, are also important prognostic factors. Patients who have a significant amount of residual disease in the resected specimen following treatment with neoadjuvant therapy have a poor prognosis compared to those with minimal or no evidence of disease in the pathological specimen [7]. Children have a better prognosis compared to adults [8]. However, the greater tumor burden in adults, use of lower doses of alkylating agents, and the timing of local therapy may explain the difference between the two groups [9]. There are multiple different structural possibilities for gene fusions in these tumors. Specific cytogenetic and molecular alterations such as p53 mutations, deletions of CDKN2A gene coding P16, P14ARF and deletion of 1p36, or all are associated with worse prognosis [10-12].
Multidisciplinary approach coupled with risk-adapted intensive neoadjuvant and adjuvant multi-agent chemotherapies along with other modalities such as radiation and surgery for control of the primary site and metastatic disease if needed. Since Ewing sarcoma family of tumors is usually rare, it does require referral to sarcoma specialists. The primary treatment will involve multidrug chemotherapy with alternating cycles of VDC and IE given every 2 weeks with growth factor support in patients with localized disease. This was based on a randomized phase 3 clinical trial where patients were randomized to the above regimen compared to VDC and dactinomycin (VDCA) 5 years event-free survival was 69% in the containing IE compared to 54% in VDCA arm. The overall survival was also significantly better, and the experimental arm containing IE versus VDCA at 72% and 61%, respectively [13]. Local control can be achieved either with surgery or radiation or both. For patients with metastatic disease, the management depends on the site and the number of metastatic lesions. Patients with limited pulmonary metastatic disease are generally treated with a curative intent to undergo multi-agent intense chemotherapy and pulmonary resection with or without radiation, with improved survival compared to other sites of metastatic disease. The addition of IE to the VDC backbone did not change patients’ outcomes with the metastatic disease; however, it showed significant improvement in nonmetastatic disease and is the commonly used regimen in the USA [13].
Patients with the relapsed disease tend to have a poor prognosis. Generally, systemic therapy involves either topotecan/cyclophosphamide or irinotecan/temozolomide [14, 15]. Surgery and radiation can be used in selective cases such as local recurrence. Vascular endothelial growth factor receptor inhibitors such as cabozantinib have shown activity in a phase 2 trial with a progression-free survival of 5 months and an overall response rate of 26% [16].
Conclusions
We described a young adult patient with locally aggressive extraskeletal Ewing sarcoma causing cord compression and paraparesis, which initially masqueraded as a local lung mass. This presentation is extremely rare and emphasizes the importance of knowing the aggressive nature of the tumor. The case also describes the challenges in management while, in this case, sepsis can prolong the initiation or continuation of management owing to the prolonged course of recovery and poor prognosis.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Both verbal and written consents were obtained.
Author Contributions
MP, VS collected and reviewed patients’ chart. MP, VS, MB, GPM and VM contributed to writing introduction, discussion and conclusion. All authors contributed equally to preparation of this manuscript and all of the authors reviewed the manuscript and agreed with the findings and interpretation.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
BMI: body mass index; ER: emergency room; VATS: video-assisted thoracoscopic surgery; PET: positron emission tomography; CD: cluster of differentiation; FLI1: friend leukemia integration 1 transcription factor; AE1/AE3: anti-cytokeratin antibody; CK: cytokeratin; SOX10: SRY-related HMG-box 10; TTF1: thyroid transcription factor; EWSR1: Ewing sarcoma breakpoint region 1; TLSO: thoracic-lumbar-sacral orthosis; PCP: Pneumocystis pneumonia
| References | ▴Top |
- Karski EE, Matthay KK, Neuhaus JM, Goldsby RE, Dubois SG. Characteristics and outcomes of patients with Ewing sarcoma over 40 years of age at diagnosis. Cancer Epidemiol. 2013;37(1):29-33.
doi pubmed - GurneyJG, Swensen AR, Bulterys M. Malignant bone tumors. In: Ries LA, Smith MA, Gurney JG, et al (Eds). Cancer incidence and survival among children and adolescents: United Stated SEER Program 1975 - 1995, NCI, SEER Program, Bethesda, MD; 1999.
- Ahmed SK, Robinson SI, Okuno SH, Rose PS, Laack NN. Adult ewing sarcoma: survival and local control outcomes in 102 patients with localized disease. Sarcoma. 2013;2013:681425.
doi pubmed - Valdes M, Nicholas G, Verma S, Asmis T. Systemic therapy outcomes in adult patients with Ewing sarcoma family of tumors. Case Rep Oncol. 2017;10(2):462-472.
doi pubmed - Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, Kovar H, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol. 2015;33(27):3036-3046.
doi pubmed - Argon A, Basaran M, Yaman F, Dizdar Y, Sakar B, Camlica H, Bavbek SE, et al. Ewing's sarcoma of the axial system in patients older than 15 years: dismal prognosis despite intensive multiagent chemotherapy and aggressive local treatment. Jpn J Clin Oncol. 2004;34(11):667-672.
doi pubmed - Cotterill SJ, Ahrens S, Paulussen M, Jurgens HF, Voute PA, Gadner H, Craft AW. Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol. 2000;18(17):3108-3114.
doi pubmed - Baldini EH, Demetri GD, Fletcher CD, Foran J, Marcus KC, Singer S. Adults with Ewing's sarcoma/primitive neuroectodermal tumor: adverse effect of older age and primary extraosseous disease on outcome. Ann Surg. 1999;230(1):79-86.
doi pubmed - Gupta AA, Pappo A, Saunders N, Hopyan S, Ferguson P, Wunder J, O'Sullivan B, et al. Clinical outcome of children and adults with localized Ewing sarcoma: impact of chemotherapy dose and timing of local therapy. Cancer. 2010;116(13):3189-3194.
doi pubmed - Hattinger CM, Rumpler S, Strehl S, Ambros IM, Zoubek A, Potschger U, Gadner H, et al. Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Genes Chromosomes Cancer. 1999;24(3):243-254.
doi - de Alava E, Antonescu CR, Panizo A, Leung D, Meyers PA, Huvos AG, Pardo-Mindan FJ, et al. Prognostic impact of P53 status in Ewing sarcoma. Cancer. 2000;89(4):783-792.
doi - Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, Healey JH, Wexler LH, et al. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J Clin Oncol. 2005;23(3):548-558.
doi pubmed - Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, Gebhardt MC, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694-701.
doi pubmed - Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jurgens H. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer. 2006;47(6):795-800.
doi pubmed - Casey DA, Wexler LH, Merchant MS, Chou AJ, Merola PR, Price AP, Meyers PA. Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer. 2009;53(6):1029-1034.
doi pubmed - Italiano A, Mir O, Mathoulin-Pelissier S, Penel N, Piperno-Neumann S, Bompas E, Chevreau C, et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020;21(3):446-455.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.