

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 11, Number 5, May 2020, pages 120-124
Successful Management of Intrathoracic Phrenic Malignant Peripheral Nerve Sheath Tumor by Multimodal Treatment
Ghizlane Raisa, d, Mehdi Maidib, Fadoua Raisc
aDepartment of Medical Oncology, Regional Center of Oncology, Agadir, Morocco
bDepartment of Thoracic Surgery, CHU AGADIR, Agadir, Morocco
cRadiotherapy Department, McGill University Cedars Cancer, Montreal, Canada
dCorresponding Author: Ghizlane Rais, Department of Medical Oncology, Regional Center of Oncology, Agadir 80322, Morocco
Manuscript submitted April 5, 2020, accepted April 13, 2020
Short title: Management of Intrathoracic Phrenic MPNST
doi: https://doi.org/10.14740/jmc3464
| Abstract | ▴Top |
Malignant peripheral nerve sheath tumors (MPNSTs) are rare and can be associated with type 1 neurofibromatosis. They are rarely located at the mediastinum, most often at the posterior mediastinal compartment or the paravertebral gutters. MPNSTs of the anterior mediastinum arising from the phrenic nerve are exceptional. Their prognosis is poor due to the high potential of local tumor recurrence and metastasis. Complete surgical resection remains the only radical treatment. However, the outcomes after this treatment alone cannot be considered satisfactory. Preoperative chemotherapy could be an interesting therapeutic option for unresectable tumors. We report here the case of a 77-year-old woman with an MPNST located at the anterior mediastinum arising from the phrenic nerve. This tumor was successfully managed with multimodal therapy combining preoperative chemotherapy, complete surgery and adjuvant radiation.
Keywords: Malignant peripheral nerve sheath tumor; Phrenic nerve; Multimodal therapy
| Introduction | ▴Top |
Malignant peripheral nerve sheath tumors (MPNSTs) are rare sarcomas which originate from peripheral nerves or from cells associated with the nerve sheath, such as Schwann cells, perineural cells, or fibroblasts [1]. They represent 5-10% of soft tissue sarcomas (STSs) and can occur either spontaneously or in association with type 1 neurofibromatosis [1, 2]. Those involving intrathoracic nerves and located at the anterior and middle mediastinum are even more exceptional [2-4]. To the best of our knowledge, few cases of MPNSTs arising from the phrenic nerve have been described in the literature [3-6].
MPNSTs are very aggressive tumors with high recurrence rate after excision and increased risk of distant metastasis. Their prognosis is poor with a 5-year overall survival of 50% [4-7]. Complete surgical resection with safe margins remains the treatment of choice. Adjuvant or neoadjuvant radiation has been proven to be a significant treatment tool in several series. Chemotherapy can be used as adjuvant treatment, but its efficacy has not been proven. It can be useful for metastatic disease or in neoadjuvant setting for large and unresectable tumors [8]. In this report, an unusual case of MPNST arising from phrenic nerve is described. A particular therapeutic approach is presented here, with relevant literature review. Multimodal treatment including preoperative chemotherapy, wide local resection with safe margins and adjuvant radiation, constitutes a successful management of this unresectable MPNST. Complete surgical resection is the best indicator of good prognosis [7].
| Case Report | ▴Top |
A 77-year-old woman presented with dyspnea, dry cough and right-sided chest pain for the previous 6 months. On physical examination, breath sounds were decreased on the right side. The skin examination showed no “cafe au lait” spots or subcutaneous tumors.
Contrast enhanced computed tomography (CT) scan of the chest revealed a well circumscribed heterogenous mass of the anterior mediastinum lateralized to the right measuring 11 cm in major axis without compression of the superior vena. This mass presents close contact with the ascending aorta and the right atrium with obvious pericardial thickening (Fig. 1). An interventional radiologist performed a transthoracic needle core biopsy, which revealed a mesenchymal lesion. An anterior mediastinotomy (Chamberlain procedure) was then performed to obtain adequate tissue for a precise diagnosis. On microscopic examination, the tumor consisted of neoplastic spindle cell proliferation arranged in interlacing bundles with large and irregular nuclei and frequent mitoses (around 10 mitoses per 10 fields) (Fig. 2a, b). Immunohistochemistry (IHC) staining was performed. The panel of monoclonal antibodies consisted of cytokeratin 5, 6, 7, and 20, vimentin, smooth muscle actin, CD-117, thyroid transcription factor-1 (TTF1), epithelial membrane antigen (EMA), CD-34, and S-100 protein. Tumor cells were positive for vimentin strongly (Fig. 3) and uniformly and negative for PS100 and other markers. This morphological and immunohistochemical aspect was compatible with high-grade MPNST. After pathological diagnosis was established, whole body CT scan was performed but did not reveal any tumor. The tumor was considered unresectable and the patient received three cycles of chemotherapy based on adriamycin 75 mg/m2. Chest CT scan performed thereafter showed partial response consisting of 30% decrease of the tumor volume allowing complete surgical excision of the tumor (Fig. 4).
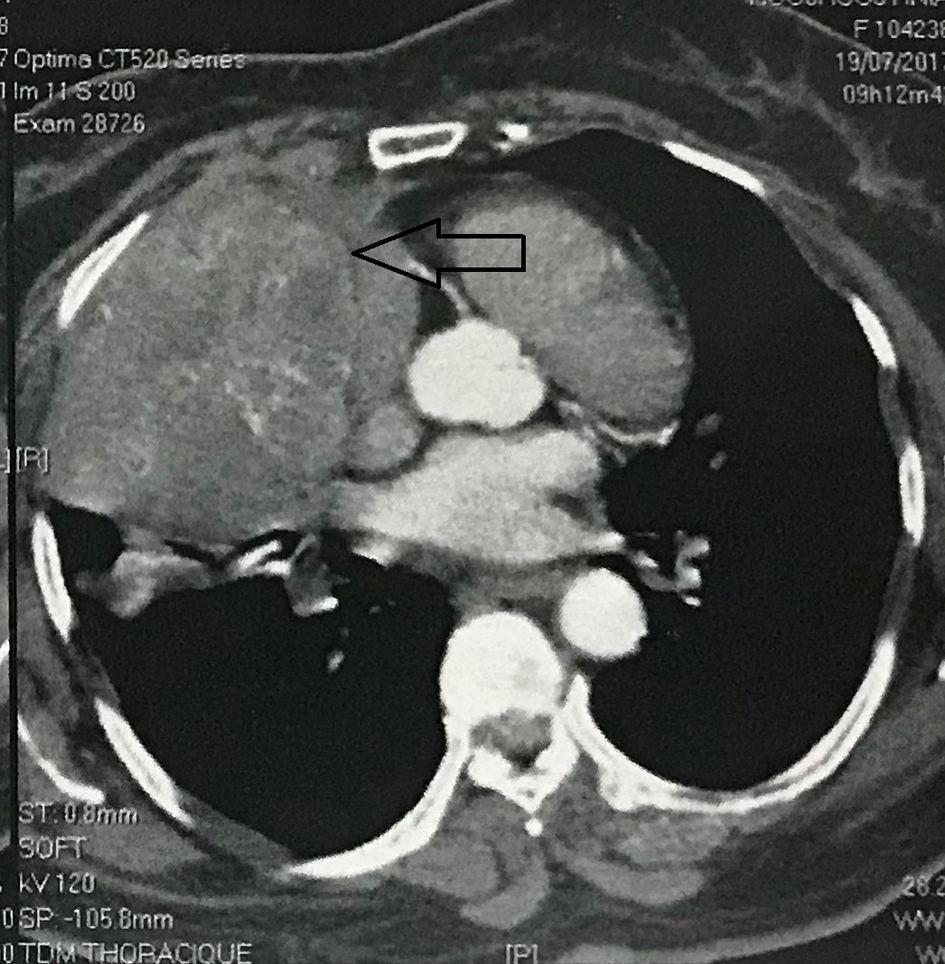 Click for large image | Figure 1. A contrast-enhanced computed tomography showing a heterogeneous mass with contrast enhancement in the right anterior mediastinum. |
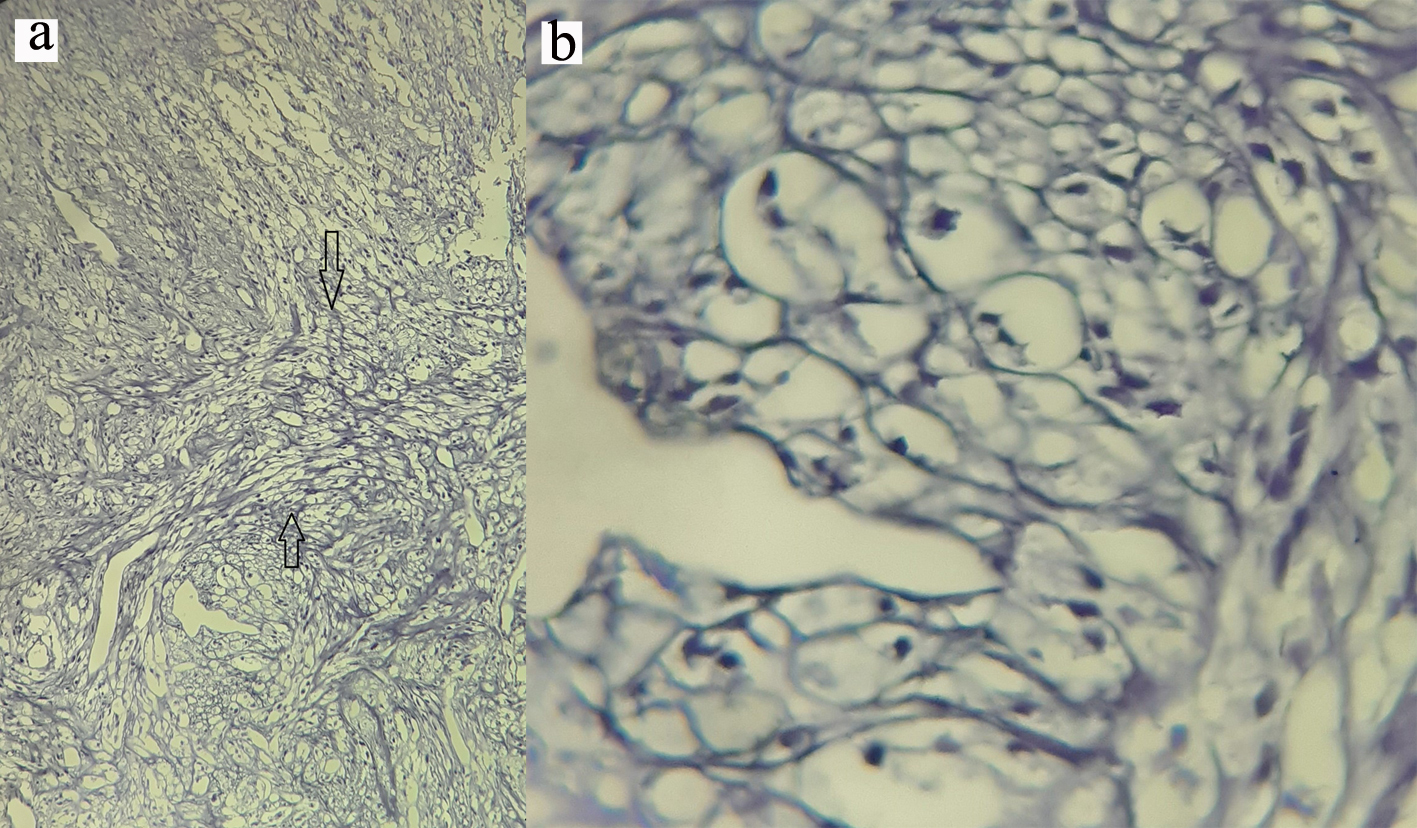 Click for large image | Figure 2. (a) Oval to spindle shaped cells with hyperchromatic and pleomorphic nuclei (magnification, × 4). (b) High mitotic activity in tumor cells (magnification, × 40). |
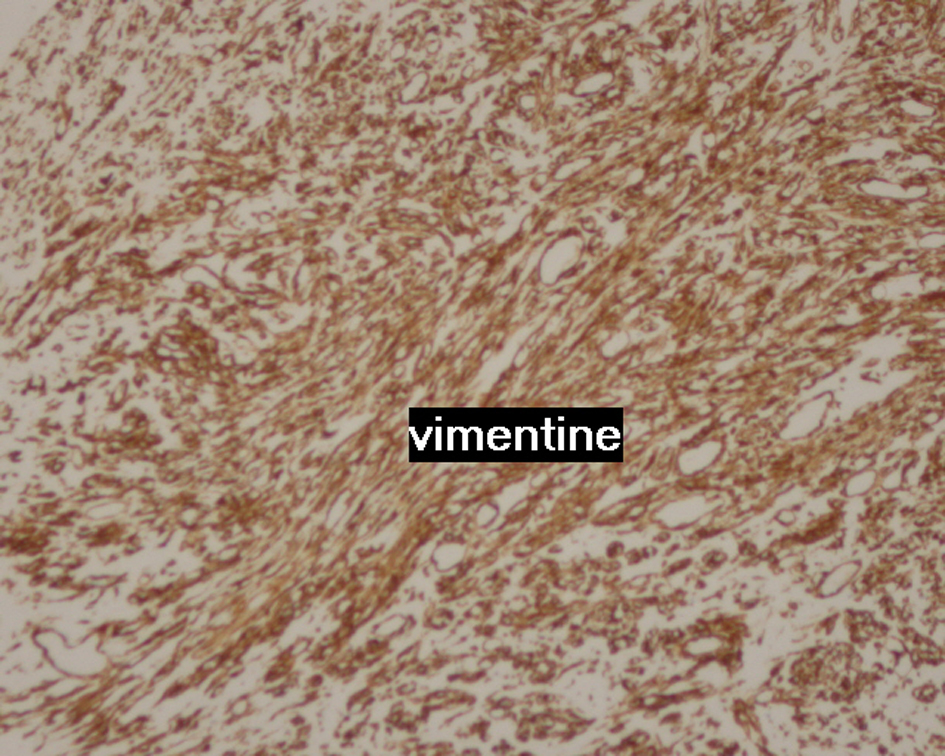 Click for large image | Figure 3. Image showing strongly and uniformly positive vimentin staining. |
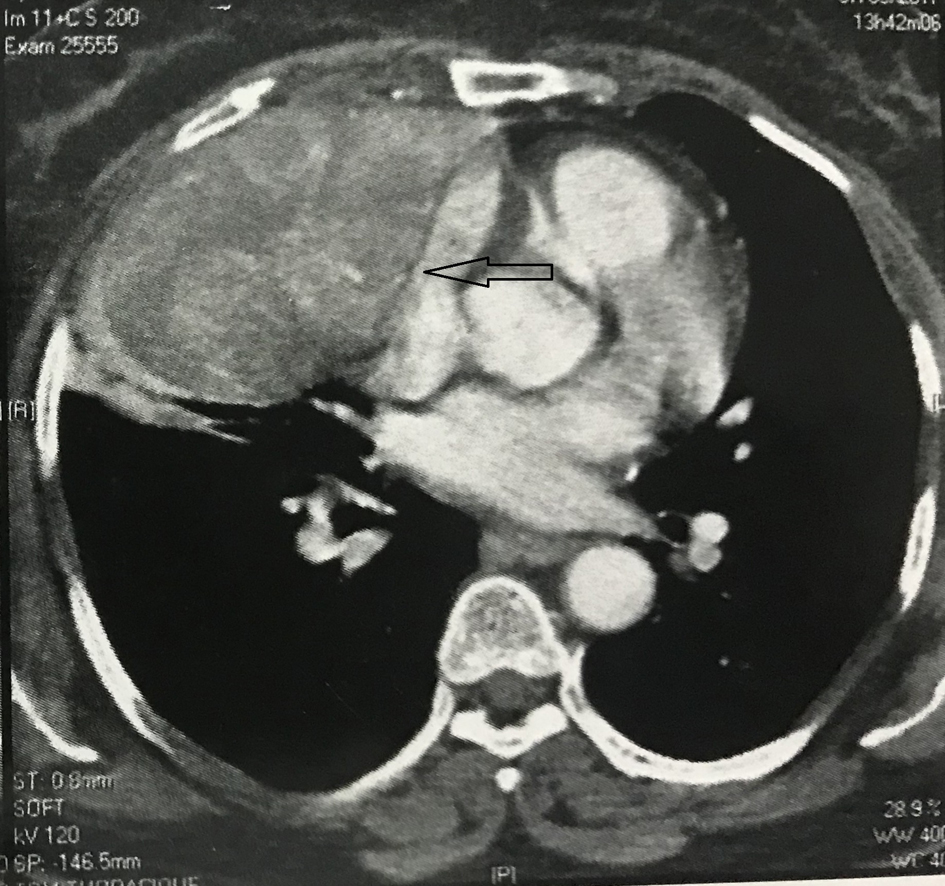 Click for large image | Figure 4. CT scan after three cycles of chemotherapy showing a partial response of 30% in the volume of the mass. CT: computed tomography. |
Right posterolateral thoracotomy was performed over the fifth rib using double-lumen endotracheal tube to permit unilateral deflation of the right lung. Surgical exploration revealed firm, irregular, and completely encapsulated mass involving the right phrenic nerve, with conservation of the cleavage plane with the trachea, the superior vena cava, the ascending thoracic aorta, and lung. The tumor was completely resected and partial resection of the right phrenic nerve was performed ensuring R0 surgery (Figs. 4, 5).
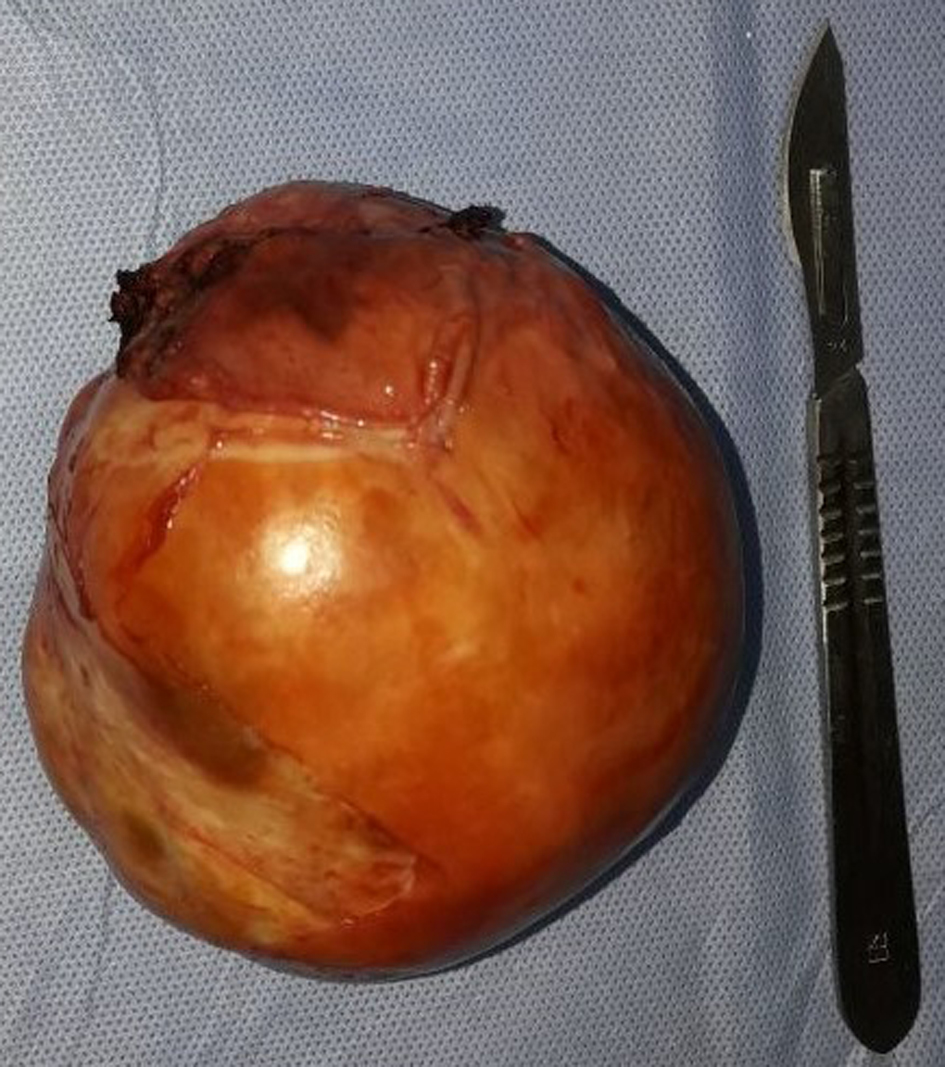 Click for large image | Figure 5. Nodular soft tissue mass measuring 10 × 9 × 6 cm and weighing 342 g. |
Final pathology confirmed the diagnosis of MPNST originating from the right intrathoracic phrenic nerve in an old female patient with no clinical evidence of Von Recklinghausen disease. No areas of hemorrhage or necrosis were seen.
The postoperative course was uneventful, and the patient was discharged on the fourth postoperative day. Six weeks later, the patient underwent adjuvant three-dimensional conformal external beam radiation to the surgical bed at the dose of 60 Gy in 30 fractions with mild acute side effects and overall a very good tolerance.
The patient remained free of recurrence 3 years after her treatment.
| Discussion | ▴Top |
There are two main reasons to present this interesting case report. First is the unusual demographics and location of MPNST in our patient [6-8]. Second one is the successful management with multimodal therapy, which suggests probable benefit of preoperative chemotherapy in some patients [9, 10].
MPNST of the intrathoracic phrenic nerve is a malign neurogenic tumor that arises from the nerve sheath and can involve the middle or anterior mediastinum. Although there have been few reports of the thoracic location of this neoplasm, most of them are located at the posterior mediastinum in the paravertebral gutters [11]. MPNSTs of the phrenic nerve have been described previously, but they appear to be even rarer than neurofibromas, with less than 20 cases reported in the English literature [12-14].
MPNSTs are typically seen in patients between 20 and 50 years old, although occasional cases may be seen in the elderly population. Known main risk factors include previous radiation exposure (either therapeutic or environmental), pre-existing neurofibromas, and neurofibromatosis type 1 (Von Recklinghausen disease), which is the most important risk factor, accounting for almost 50% of patients with MPNST [10]. In our case, the patient is female, 77 years old. The site of the injury is the phrenic nerve and the patient has no sign of neurofibromatosis-1.
Diagnosis is particularly difficult because of the variable pathological presentation and unpredictable differentiation. The clinical features depend on the site, size, and malignant potential of the tumor [10, 11]. Most of these tumors occurring inside the chest or abdomen take longer for the symptoms to express and in many cases discovery is delayed, meaning that the tumor grows and in many cases can no longer be completely excised upon discovery, or may be inoperable. These tumors are often found incidentally at standard chest X-ray [15]. Phrenic nerve tumors should be suggested in front of any rounded mass opposite the path of the nerve and should not be confused with primary cardiac tumors. In our case, there were some symptoms such as chest pain, cough, and dyspnea related to the mass effect.
The microscopic diagnosis of MPNSTs is difficult because of their cellular origin and histopathological similarities with other spindle cell sarcomas like monophasic synovial sarcoma, leiomyosarcoma and fibrosarcoma [13]. Thus, a combination of gross, histopathological, and immunohistochemical studies are used for diagnosing these tumors. Malignancy is retained on cell pleomorphism, high number of mitosis and capsular infiltration [13-15]. Also, the lack of well-delimited borders and encapsulation, invasion signs, significant atypia and necrosis suggest malignancy. Large areas of necrosis and high mitotic activity are typical [13].
IHC guides the diagnosis but is not specific. In contrast to benign and locally aggressive schwannomas, which demonstrate uniform immunoreactivity for S-100 protein, malignant entity may show patchy or absent immunoreactivity for this antigen [10-13]. In our case, IHC showed intense positivity to vimentin and negativity to PS100. Diagnosis was based on both morphological and immunohistochemical aspects which were compatible with high-grade MPNST.
Full assessment is required before starting treatment. The minimum requirements for staging include chest imaging by CT scan and/or magnetic resonance imaging (MRI), which are necessary to localize the tumor accurately, and to provide information about tumor size and involvement of adjacent organs, and help in surgical planning. Although there is significant overlap between imaging features of large benign and malignant peripheral nerve sheath tumors. MPNST should be suspected if there is heterogeneous density, focal areas of central necrosis, ill-defined borders, rapid growth on interval imaging, invasion of surrounding soft tissues, and destruction of surrounding bone structures [13, 14]. MPNSTs have metastatic potential, which requires distant workup staging [11]. Metastatic spread is hematogenous and metastatic locations are diverse. In our case, the antero-inferior mediastinal tumor appears to be of inhomogeneous density with enhancement of the periphery and the central portion without any detected metastasis.
Prognostic factors are tumor size, grade of malignancy, speed of clinical evolution, degree of cellular pleomorphism and mitotic activity. Prognosis is usually poor, especially in large invasive tumors, those related to NF1, and those with high-grade histologic features [8, 12-16].
The mainstay of therapy of MPNST is complete surgical resection with negative margins [5, 12-16]. MPNSTs are biologically aggressive in nature with rapid progression, and then surgery must be planned as soon as the pathological diagnosis has been made. Moreover, surgery should preferably be performed by specialized surgeons in the management of sarcomas; this could ensure higher rates of complete surgical resection which is the best indicator of good prognosis. Currently the introduction into clinical practice of additional chemotherapy and radiotherapy can improve immediate and long-term treatment results of such patients [8, 17].
The use of pre- or postoperative adjuvant therapy is not yet well codified. Adjuvant radiotherapy to the tumor bed and possibly other sites of bulky may be beneficial in preventing local tumor relapse. It may be a very useful therapy to improve local control [10-12, 15]. In our patient, MPNST was identified in the anterior mediastinum extending behind the right anterior chest wall with suspicious invasion of its surrounding capsule. Complete wide excision of the mass was done with parietal resection and finally negative margins were obtained. Adjuvant radiotherapy to the tumor bed was indicated to ensure local control and to decrease any chance of further relapse. Our patient is still asymptomatic with no evidence of recurrence so far.
Recurrences of MPNST are associated with poor prognosis. Zou et al reported that MPNST recurrences are strongly associated with positive surgical margins, poorly sensitive to chemotherapy and relatively more radioresistant [15].
There are few data regarding the efficacy of chemotherapy on MPNSTs. Neoadjuvant chemotherapy could be considered in case of unresectable disease, or when tumor excision requires mutilating surgery. It is based on anthracycline-based chemotherapy, whether or not combined with external radiotherapy, possibly in concomitant treatment.
The role of chemotherapy in advanced MPNST is unclear. Most MPNSTs are biologically high-grade sarcomas that tend to recur (40-65%) and metastasize (40-80%) most commonly to the lungs [8, 10, 17]. A retrospective study compared the efficacy of first-line chemotherapy based on anthracyclines and/or ifosfamide [18] in patients with MPNST (175 patients) with those with other types of STSs (2,500 patients). This study did not show any difference between the two groups in terms of objective response (21% and 22% respectively), progression-free survival (17 and 16 weeks), or overall survival (48 and 51 weeks). However, treatment with combination of doxorubicin and ifosfamide achieved higher response rate (not specified in the article) than that obtained with doxorubicin alone, but without overall survival improvement.
Only one prospective phase II study specifically looked at the efficacy of chemotherapy in patients with metastatic MPNST [19]. Chemotherapy consisted of two cycles of a combination with adriamycin/ifosfamide, followed by two cycles of combination with etoposide/ifosfamide. Forty-eight patients (33 MPNSTs on NF1 and 15 sporadic) were included. This treatment produced a response rate of 17.2% for patients with NF1 and 33.3% for sporadics. In addition, this study highlights the potential role of etoposide in the treatment of MPNST.
Conclusions
The addition of our case to the literature offers new clinicopathological data useful for better defining the diagnosis and biological behavior of MPNST. The purpose of presenting this case is not only to report an uncommon tumor but also to raise awareness among clinicians adding this clinical entity as a differential diagnosis when a mediastinal anterior mass is identified. Neoadjuvant chemotherapy or radiotherapy should be considered in locally advanced disease to obtain complete resection. Better knowledge of these tumors, particularly of their molecular origins, should allow their classification and therefore their treatment to evolve.
Acknowledgments
The authors gratefully acknowledge the reference expertise of the Youssef Bouhajeb from Ibn Rochd Histopathology Center, Agadir, Morocco.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Author Contributions
All authors contributed to the preparation of this manuscript, and interpretation of the case. All the authors have read and approve the final version of the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Hruban RH, Shiu MH, Senie RT, Woodruff JM. Malignant peripheralnerve sheath tumors of the buttock and lower extremity. A study of 43 cases. Cancer. 1990;66:1253-1265.
doi - Saito H, Minamiya Y, Tozawa K, Matsuzaki I, Taguchi K, Nakagawa T, Ogawa J. Mediastinal neurofibroma originating from the left intrathoracic phrenic nerve: report of a case. Surg Today. 2004;34(11):950-953.
doi pubmed - Le Pimpec-Barthes F, Martinod E, Riquet M, Saint-Blancard P, Jancovici R. [Tumors of the phrenic nerve]. Rev Mal Respir. 1998;15(1):93-95.
- Azarow KS, Pearl RH, Zurcher R, Edwards FH, Cohen AJ. Primary mediastinal masses. A comparison of adult and pediatric populations. J Thorac Cardiovasc Surg. 1993;106(1):67-72.
doi - Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021.
doi - Bacha S, Chaouch N, Ayadi A, Zidi A, Cheikhrouhou S, Racil H, Chabbou A. Tumeur maligne des gaines des nerfs peripheriques: masse exceptionnelle du mediastin anterieur et moyen. RevPneumol Clin. 2015;71(6):364-368.
doi pubmed - Mordant P, Le Pimpec-Barthes F, Riquet M. [Neurogenic tumors of the mediastinum in adults]. Rev Pneumol Clin. 2010;66(1):81-94.
doi pubmed - Shimoyama T, Yoshiya K, Yamato Y, Koike T, Honma K. Long-term survival after removal of a malignant peripheral nerve sheath tumor originating in the anterior mediastinum. Gen Thorac Cardiovasc Surg. 2009;57(6):310-314.
doi pubmed - Stucky CC, Johnson KN, Gray RJ, Pockaj BA, Ocal IT, Rose PS, Wasif N. Malignant peripheral nerve sheath tumors (MPNST): the Mayo Clinic experience. Ann Surg Oncol. 2012;19(3):878-885.
doi pubmed - Rais F, Benhmidou N, Rais G, Kouhen F, Bellahamou K, Loughlimi H, Maghous A, et al. Solitary intraosseous schwannoma of the base and vault of the skull: a summary review of such unusual location. Clin Sarcoma Res. 2015;5:6.
doi pubmed - Koezuka S, Hata Y, Sato F, Otsuka H, Makino T, Tochigi N, Iyoda A. Malignant peripheral nerve sheath tumor in the anterior mediastinum: A case report. Mol Clin Oncol. 2014;2(6):987-990.
doi pubmed - Nakajima Y, Mikami I, Akiyama H, Kinoshita H, Iijima Y, Uramoto H. Long-term progress of six cases of malignant peripheral nerve sheath tumors of the mediastinum that underwent surgical treatment: Case report series. Int J Surg Case Rep. 2016;24:185-187.
doi pubmed - Ralli M, Singh S, Hasija S, Verma R. Intrathoracic malignant peripheral nerve sheath tumor: histopathological and immunohistochemical features. Iran J Pathol. 2015;10(1):74-78.
- Kamran SC, Shinagare AB, Howard SA, Nishino M, Hornick JL, Krajewski KM, Ramaiya NH. Intrathoracic malignant peripheral nerve sheath tumors: imaging features and implications for management. Radiol Oncol. 2013;47(3):230-238.
doi pubmed - Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang WL, Zhang W, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014-1022.
doi pubmed - Wong WW, Hirose T, Scheithauer BW, Schild SE, Gunderson LL. Malignant peripheral nerve sheath tumor: analysis of treatment outcome. Int J Radiat Oncol Biol Phys. 1998;42(2):351-360.
doi - Kroep JR, Ouali M, Gelderblom H, Le Cesne A, Dekker TJ, Van Glabbeke M, Hogendoorn PC, et al. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: an EORTC soft tissue and bone sarcoma group study. Ann Oncol. 2011;22(1):207-214.
doi pubmed - Kolberg M, Holand M, Agesen TH, Brekke HR, Liestol K, Hall KS, Mertens F, et al. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol. 2013;15(2):135-147.
doi pubmed - Widemann B, Reinke DK, Helman LJ, Ludwig JA, Schuetze S, Staddon AP, Milhem MM, et al. SARC006: Phase II trial of chemotherapy in sporadic and neurofibromatosis type 1 (NF1)-associated high-grade malignant peripheral nerve sheath tumors. ASCO Annual meeting. 2013. Abstract 10522.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.