

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 10, Number 7, July 2019, pages 218-221
Lymphangioleiomyomatosis: A Challenging Case
Joana Serodioa, e, Joana Carneiroa, Helena Vilacab, Manuel Veigac, Ivone Goncalvesd
aInternal Medicine Department, Unidade Local de Saude do Alto Minho, Ponte de Lima, Portugal
bInternal Medicine Department, Centro Hospitalar Tamega e Sousa, Penafiel, Portugal
cAnatomical Pathology Department, Unidade Local de Saude do Alto Minho, Viana do Castelo, Portugal
dPneumology Department, Unidade Local de Saude do Alto Minho, Viana do Castelo, Portugal
eCorresponding Author: Joana Serodio, Internal Medicine Department, Unidade Local de Saude do Alto Minho, Largo Conde de Bertiandos, Ponte de Lima 4990-041, Portugal
Manuscript submitted July 3, 2019, accepted July 25, 2019
Short title: Lymphangioleiomyomatosis
doi: https://doi.org/10.14740/jmc3329
| Abstract | ▴Top |
Lymphangioleiomyomatosis (LAM) is a rare progressive cystic lung disease, most prevalent in women, which affects the lung function, axial lymphatics and causes angiomyolipomas. Progressive dyspnea, pneumothorax, chylous effusions and cough are some of the clinical manifestations. It is a disease that can be difficult to manage and can have a poor prognosis, especially if not promptly addressed. We present a case report of a 43-year-old woman who was admitted in 2014 with recurrent chylothorax and diagnosed with LAM. However, since the actual best therapeutic approach, sirolimus, was not yet recommended at the time, together with the patient’s low adherence to medical follow-up, her clinical management was difficult with some complications. After her return into medical care, the disease had progressed substantially; however, it stabilized after starting sirolimus therapy. This case report aims to exemplify how the management of this disease can be difficult and highlight sirolimus as the “weapon” to improve its prognosis even in an advanced stage of the disease.
Keywords: Lymphangioleiomyomatosis; Chylous pleural effusion; Cystic lung disease; Sirolimus
| Introduction | ▴Top |
Lymphangioleiomyomatosis (LAM) is a rare progressive cystic lung disease that usually affects women of child-bearing age [1]. It can occur sporadically or be associated with tuberous sclerosis complex (TSC) and it is caused by infiltration of the lung with smooth muscle-like cells (LAM cells) that arise from an unknown source and contain growth-activating mutations in tuberous sclerosis genes [2]. They spread via blood and lymphatics and result in cystic changes in the lungs and axial lymphatics (lymphangioleiomyomas), and abdominal tumors called angiomyolipomas (AMLs) mostly present in the kidneys [2, 3].
Progressive dyspnea and pneumothorax that commonly recur are the main clinical manifestations, followed by chylous effusions (pleural or abdominal), cough, hemoptysis and chyloptysis, and abdominal hemorrhage caused by AMLs [3].
It is a disease that can be difficult to manage and can have a poor prognosis, especially if not promptly addressed [2].
| Case Report | ▴Top |
We present a non-smoking 43-year old woman, living in the countryside, with low education level, and with a history of admission in 2009 due to a recurrent right chylothorax that needed decortication, and mediastinal lesions. She was discharged to the outpatient clinic to continue investigation, but after a few months she missed the follow-up appointments.
She was admitted to the internal medicine ward in 2014 with a few weeks’ history of isolated progressive dyspnea. On physical examination she was eupnoeic, hemodynamically stable, without fever, and diminished respiratory sounds on the inferior half of the left hemithorax. Pleural effusion was demonstrated on the chest radiograph and the thoracentesis showed a fluid with chylous appearance, confirmed by its analysis with high triglycerides (1,764 mg/dL), lymphocyte predominance (86%), and an exudate according to the Light’s criteria, in a patient with a normal diet.
Revision of her previous admission indicated that she had been submitted to a biopsy of the mediastinal lesion that showed areas with vascular structures with perivascular epithelioid-like cells, and positivity for the HMB45 in the immunohistochemical study, which is compatible with LAM cells (Fig. 1).
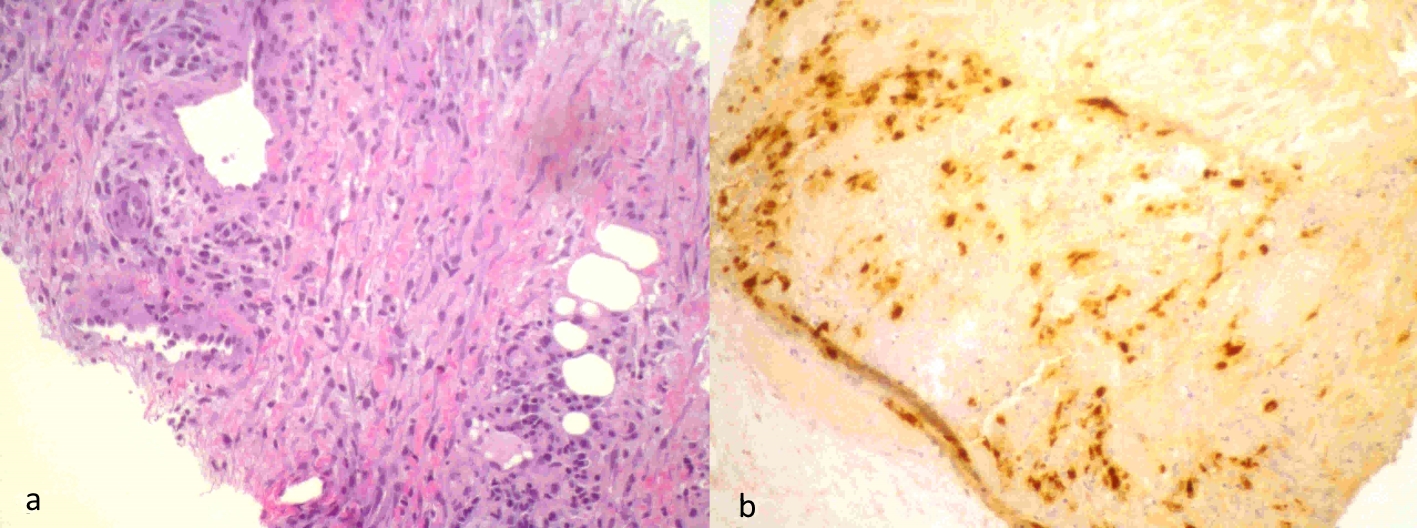 Click for large image | Figure 1. Mediastinal lesion biopsy: areas with vascular structures with perivascular epithelioid-like cells (a), which showed positivity for the HMB45 in the immunohistochemical study compatible with LAM cells (b). |
The abdominal computed tomography (CT) scan showed a left pararenal lesion that corresponded to an AML. Her first high-resolution chest CT (HRCT) scan was performed and displayed multiple bilateral thin-walled cysts (Fig. 2), making the definitive diagnosis of LAM together with chylothorax, biopsy findings of LAM cells and the pararenal AML. Furthermore, she was diagnosed with sporadic LAM after a negative evaluation for the concomitant presence of TSC. Her pulmonary function tests (PFT) showed reduced forced expiratory volume (FEV 1:40.4%), a residual volume of 109% and a reduced diffusing capacity of the lungs for carbon monoxide (DLCO) of 45.1% (Table 1). She desaturated to an SpO2 of 89% in the six-minute walk test (SMWT) but had a partial pressure of arterial oxygen (PaO2) at rest of 71 mm Hg without oxygen supplementation.
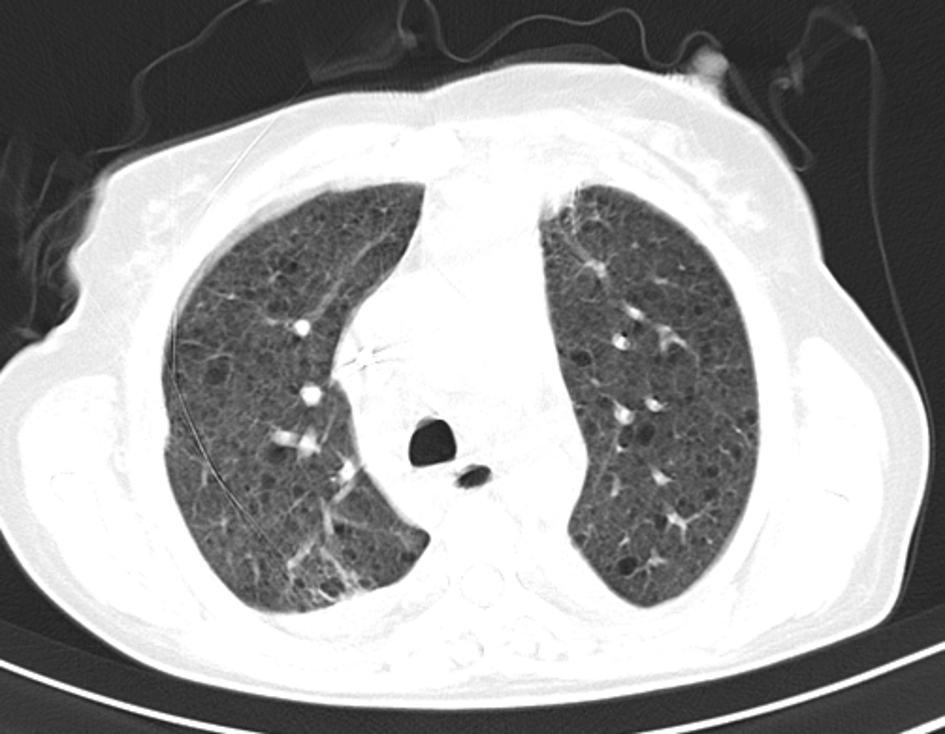 Click for large image | Figure 2. HRCT scan in 2014 with multiple thin-walled cyst nodes. |
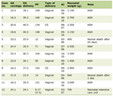 Click to view | Table 1. Pulmonary Function Tests and Clinical Follow-Up |
She rapidly evolved with recurrent chylous effusion with a first approach of intermittent therapeutic thoracentesis and the implementation of a fat-free diet together with oral supplementation of medium-sized triglycerides. Since she showed no reduction in recurrence, and after discussion with the pneumology team, she was proposed for a complete effusion drainage with a chest tube while on a fat-free total parenteral nutrition followed by pleurodesis. This was complicated with an iatrogenic pneumothorax after the chest tube insertion. Later she developed a nosocomial respiratory and pleural infection with bacteremia due to methicillin-susceptible Staphylococcus aureus, which then led to some loculation of the effusion and pleural adhesions, and a slow but gradual resolution of the pleural effusion after antibiotic treatment and fluid drainage. She was discharged to outpatient follow-up continuing the kinesiotherapy and bronchodilator therapy.
After 5 months of follow-up she once again missed the follow-up appointments, returning later in 2016 with a worsening dyspnea to mild efforts. She showed a decline in her PFTs with obstruction, FEV 1 of 44.3% and DLCO of 43.9%, together with desaturation on the SMWT to 82%, a PaO2 at rest of 64 mm Hg (Table 1), and evident disease progression on the HRCT scan with a higher number of cysts, marked fibrotic parenchymal changes, and multiple mediastinal adenopathies (Fig. 3). Ambulatory continuous oxygen supplementation was prescribed, and she was proposed for treatment with sirolimus according to the new recommendations. She started it with 2 mg once daily with a target serum level 5 - 10 ng/mL. Sirolimus was well tolerated without relevant side effects.
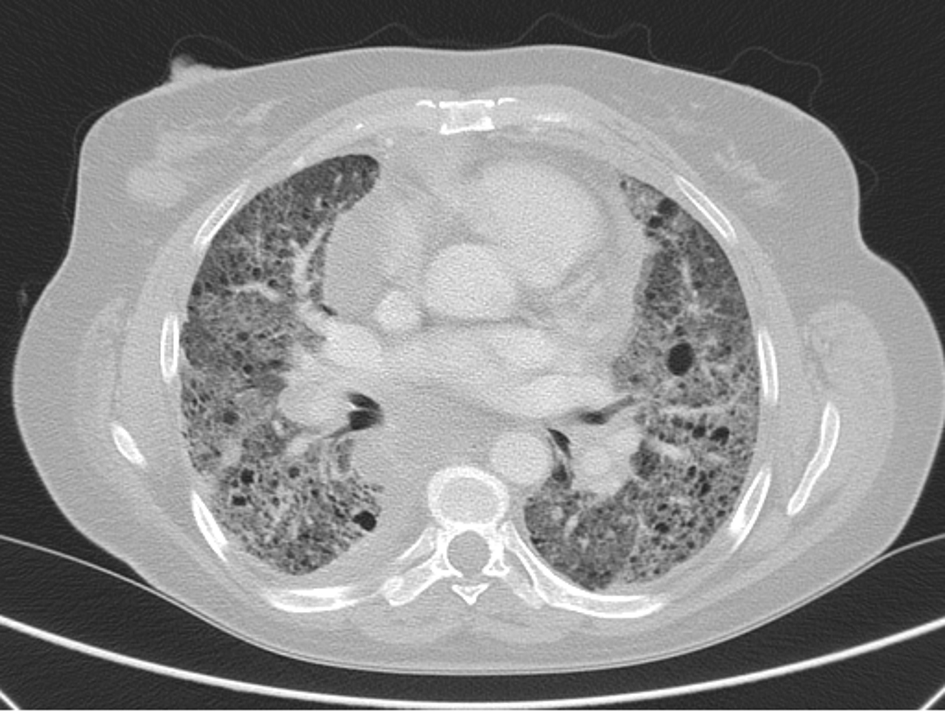 Click for large image | Figure 3. HRCT scan in 2017 showing disease progression with parenchymal fibrosis, and higher number of cysts and mediastinal adenopathies. |
She was discharged with dyspnea to minimal efforts, a PaO2 79 mm Hg with oxygen supplementation of 2L/min. During follow-up evaluations there was clinical improvement of dyspnea, with an imaging stabilization and a resolution of hypoxemia. Regarding the PFTs, there was stabilization (Table 1).
| Discussion | ▴Top |
LAM is a rare disease that might have unremarkable clinical manifestations. When approaching multi-organ pathologies, both past and present detailed clinical findings need to be taken into consideration, as in this case, to arrive at a correct diagnosis in the most expeditious manner possible.
During the last decade LAM has undergone some crucial changes in the way we look at the disease and its management. What was first classified as an interstitial lung disease is now considered a low progressive neoplastic disease that can metastasize [1, 2]. Also, sirolimus has emerged as the revolutionary therapy in order to stabilize the disease in patients with declining PFT and is also used as a first-line treatment option in the chylothorax management, before any other invasive management [4]. In this case, both times that the patient presented with recurrent chylothorax, sirolimus had not yet been proposed as a standard of care, although there was already some evidence about it but without definite conclusions. The chylous effusion was then managed according to the classical recommendations. After her return in 2016, she had a marked progression of the disease that was now very severe with hypoxemia, worsening of PFT and higher number of both pulmonary cysts and mediastinal lymph node involvement. She was still proposed to start sirolimus with an overall good outcome as shown by the improvement of dyspnea, hypoxemia, and an imaging and pulmonary function stabilization, which was consistent with the Multicenter International LAM Efficacy and Safety of Sirolimus (MILES) trial results [5]. A reduction in size of mediastinal adenopathies and a slight reduction of the parenchymal ground glass opacities in the inferior lobes were also demonstrated, showing efficacy of sirolimus even in severely advanced LAM.
This case report aims to show sirolimus efficacy even in advance stages of LAM and also to show the difficult task of dealing with real life patients who are poorly adherent and repeatedly abandon the follow-up. These factors in conjunction with having a rare disease with poor prognosis, clinical manifestations that can be difficult to manage (recurrent chylothorax), and complications of the disease management itself (nosocomial infections and iatrogenic pneumothorax) can make the treatment of this kind of patients a true challenge.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors have no conflict of interest to declare.
Informed Consent
Informed consent was obtained from the patient.
Author Contributions
JS contributed to manuscript design and writing, and was the physician that worked with the patient; JC was involved in manuscript review, and was the physician that worked with the patient; HV and MV contributed to data collection and manuscript review; IG was the attending physician, and contributed to discussion design and manuscript review.
| References | ▴Top |
- McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, Steagall WK, et al. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am J Respir Crit Care Med. 2016;194(6):748-761.
doi pubmed - Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse Cystic Lung Disease. Part I. Am J Respir Crit Care Med. 2015;191(12):1354-1366.
doi pubmed - Harari S, Torre O, Moss J. Lymphangioleiomyomatosis: what do we know and what are we looking for? Eur Respir Rev. 2011;20(119):34-44.
doi pubmed - Gupta N, Finlay GA, Kotloff RM, Strange C, Wilson KC, Young LR, Taveira-DaSilva AM, et al. Lymphangioleiomyomatosis diagnosis and management: high-resolution chest computed tomography, transbronchial lung biopsy, and pleural disease management. An Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2017;196(10):1337-1348.
doi pubmed - McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364(17):1595-1606.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.