

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 9, Number 10, October 2018, pages 348-351
Sclerosing Spindle Cell Preauricular Rhabdomyosarcoma in a 7-Year-Old Female
Niki Parikha, b, Winslo Idiculaa, Joehassin Corderoa
aTexas Tech University Health Sciences Center School of Medicine, 3601 4th Street, Lubbock, TX 79430, USA
bCorresponding Author: Niki Parikh, TTUHSC Department of Otolaryngology, 3601 4th Street, STOP 8312, Lubbock, TX 79430-8312, USA
Manuscript submitted September 17, 2018, accepted September 24, 2018
Short title: Spindle Cell Embryonal Preauricular RMS
doi: https://doi.org/10.14740/jmc3164
| Abstract | ▴Top |
Rhabdomyosarcoma (RMS) is the most common soft tissue malignancy seen in children. Approximately half of all RMS seen in the pediatric population occur in the head and neck; it is the most common malignant tumor of the aural region in childhood. There are three histological subtypes: embryonal, alveolar and pleomorphic. Sclerosing spindle cell rhabdomyosarcoma (ssRMS) is subtype of the embryonal RMS seen mostly in adult males. Although embryonal RMS is common, the spindle cell variant is rare comprising only 3% of cases and is usually seen in males below the age of 7 years. This case study demonstrates sclerosing spindle cell RMS in a 7-year-old female patient presenting with an auricular mass.
Keywords: Rhabdomyosarcoma; Auricle; Spindle cell; Soft tissue tumor; Pediatrics
| Introduction | ▴Top |
Rhabdomyosarcoma (RMS) is a primary malignancy in children and adolescents that arises from embryonic mesenchymal cells which develop into skeletal muscle [1]. RMS comprises over 50% of all soft tissue sarcomas in children, and is thus the most common soft tissue sarcoma [2]. RMS has different locations and types based on the age group. Sarcomas in adults arise mostly in the extremities, whereas RMS in children occurs in any anatomical location of the body where there is skeletal muscle as well as at sites where skeletal muscle is absent, such as in bile duct or urinary bladder [3, 4]. The disease can arise at any site and in any tissue in the body except bone. According to National Cancer Institute data, the most common sites in children are the head and neck (25%) [5], and genitourinary tract (22%), with 18% occurrence in the extremities [6]. Multimodal therapy is a contemporaneous approach that includes surgery, chemotherapy and radiation treatment [7, 8]. The embryonal type has the best prognosis with a 10-year survival of more than 80% [4]. According to a search performed on PubMed, there are no published articles of pediatric patients with sclerosing spindle cell preauricular RMS.
| Case Report | ▴Top |
A 7-year-old female was seen by her PCP when her mother noticed a non-tender swelling on her left ear that was enlarging in size. The PCP initially suspected perichondritis and the patient was prescribed a course of Levaquin; however, due to a lack of improvement, the patient was referred to an ear, nose and throat (ENT) surgeon. The ENT surgeon felt that it was not an infectious cause and discontinued Levaquin; a 7-day course of steroids was given. With no improvement, the patient was seen by the university ENT surgeon on January 21, 2008 and an initial working diagnosis of auricular pseudocyst was made. The lesion was excised in early February of 2018 and pathology showed a high-grade sarcoma with positive margins. She then underwent a PET, MRI and CT scan. The PET scan showed enhancement around the left external auditory canal and jugular chain lymphadenopathy bilaterally. The MRI also showed enhanced superficial soft tissue intensity around the left external auditory canal consistent with a history of auricular sarcoma and the CT scan was also negative (Fig. 1).
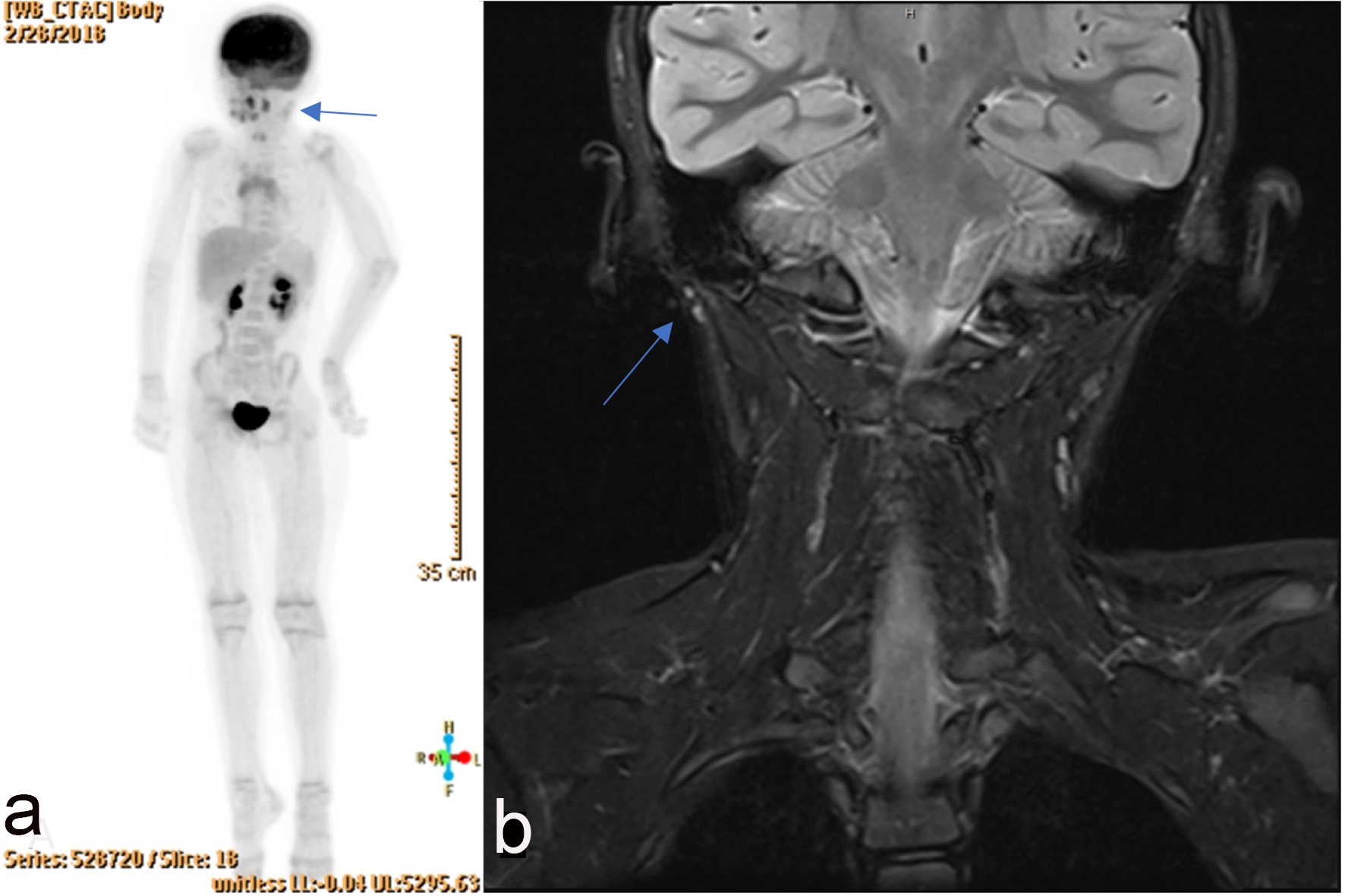 Click for large image | Figure 1. (a) PET scan with arrow pointing to enhancement around left external auditory canal. (b) MRI with arrow showing enhanced superficial soft tissue intensity around left external auditory canal. |
After the initial work-up, the patient underwent a second surgery for wider excision, sentinel lymph node biopsy, bone marrow biopsy and placement of venous access port for future chemotherapy. Lymphoscintigraphy revealed two to three nodes showing activity in the left neck which were to be removed along with excision of the sarcomatous lesion (Fig. 2). Bone marrow flow cytometry FW18-177 was performed to assess the presence of metastatic cells in bone marrow. Bone marrow biopsy showed no evidence of metastatic disease, granulomata or lymphoid aggregates. Cytogenetics was also normal; however, the sentinel lymph node was positive for malignancy.
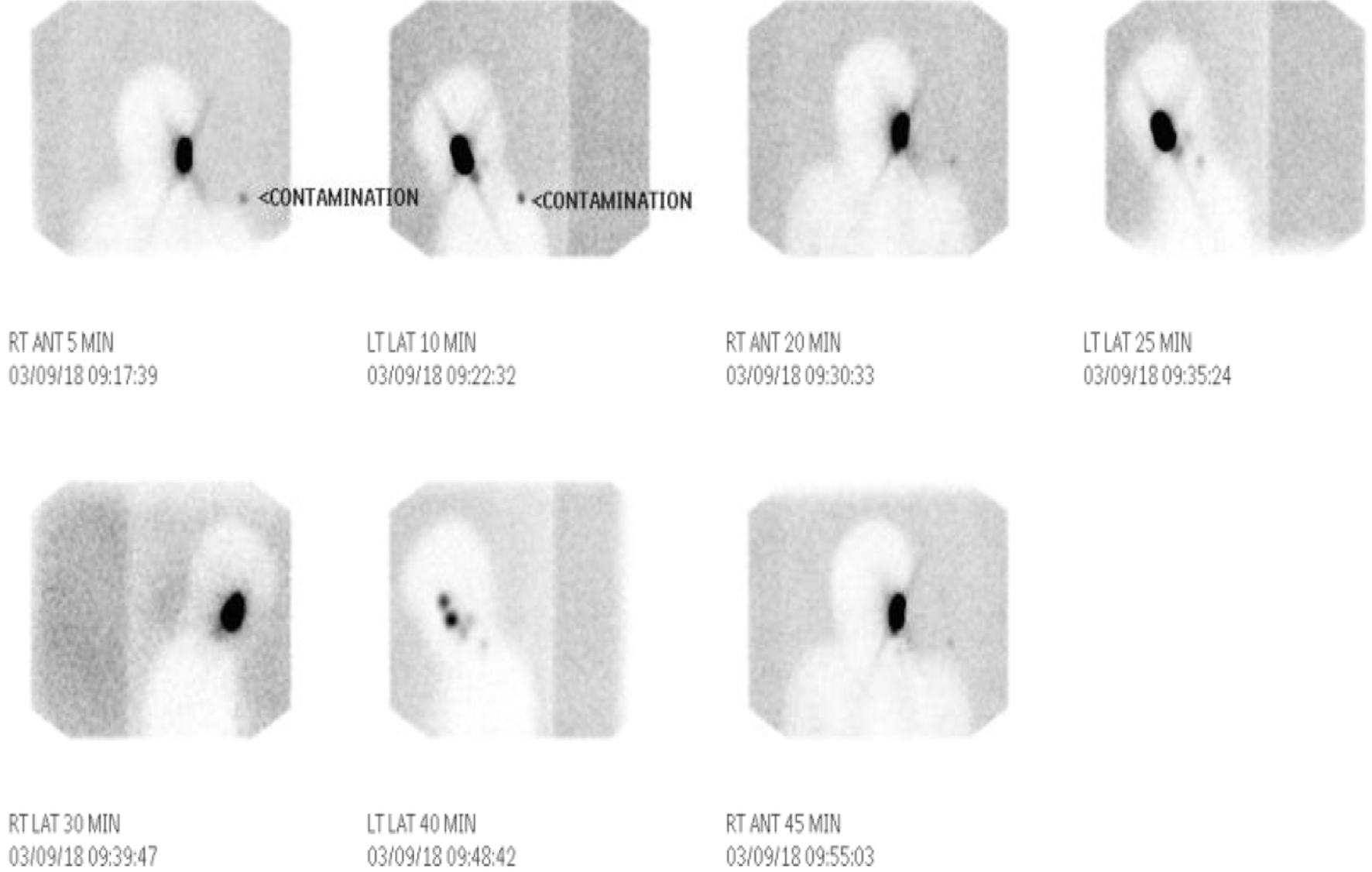 Click for large image | Figure 2. Lymphoscintigraphy revealing two to three nodes showing activity in the left neck. |
In early April 2018, she underwent re-excision of residual microscopic disease of left ear RMS. The pathological classification at this time was low-grade sarcoma. She was then referred to a children’s hospital for evaluation and was started her on a course of 24 chemotherapy cycles followed by radiation treatment. Her chemotherapy regimen consisted of vincristine, dactinomycin and cyclophosphamide with MESNA. She was later seen at the ENT clinic while she was undergoing chemo-radiation treatment and the wound was healing well without any complication (Fig. 3). The final pathological diagnosis was spindle cell sclerosing RMS (ssRMS). It is recognized by World Health Organization as a newly identified and stand-alone type.
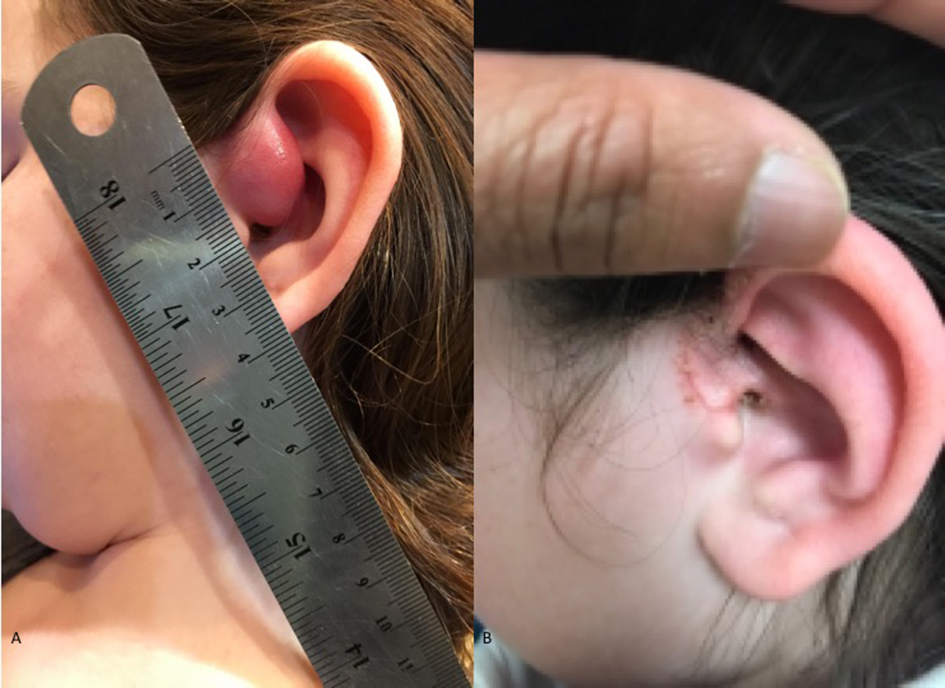 Click for large image | Figure 3. Presentation of rhabdomyosarcoma before (a) and after (b) surgery. |
| Discussion | ▴Top |
RMS is a cancerous tumor that begins in the rhabdomyoblasts, cells that differentiate into skeletal muscle. Although rare, this type of malignancy is most commonly seen in children under the age of 10 years [9]. The cancer is most common in boys and the most common locations include head and neck, urinary and reproductive organs, chest, abdomen and extremities [4]. RMS has an overall survival rate of 70%, depending on the site, and has a survival rate as high as 90% in the orbital and neck regions [4, 6]. The head and neck location is the most common site for RMS and accounts for 35-50% of tumors. Rapid diagnosis and recognition of this rare disorder will facilitate long-term survival. However, ssRMS is commonly seen with the mean age at diagnosis of about 27 years and for alveolar RMS type is about 16 years and embryonal RMS is peaking at 7 years of age [10].
There are primarily three cell types of RMS, embryonal, alveolar and pleomorphic. Alveolar is more aggressive, growing rapidly and more likely to metastasize. Embryonal RMS is the most common tumor type, comprising up to 80% of RMS malignancies, and is seen in children with an average age of 6 years. The embryonal type is subdivided into three histological subtypes. The botryoidal type is usually seen in mucosal sites and has a characteristic grape-like gross appearance. The spindle type consists of cells that have spindle morphology and is most commonly located in the orbital and para-testicular areas. Lastly the rare pleomorphic subtype is most commonly seen in extremity skeletal muscle in patient over the age of 45 years [2, 11]. Microscopically, they are 1) round cells with eccentric eosinophilic cytoplasm, 2) spindle cell myoblasts with tapered fibrillary eosinophilic cytoplasm, and 3) anaplastic [1].
RMS is occasionally seen in patients with other genetic diseases such as Li-Fraumeni syndrome, Neurofibromatosis, Beckwith-Wiedemann syndrome and Costello syndrome [9, 10, 11]. Symptoms and signs depend on tumor location [2]. There are primarily two histological types of RMS with genetic mutations that play a role in pathogenesis of this malignancy, the alveolar subtype and the embryonal subtype. The alveolar type a translocation between long arm of chromosome 2 and chromosome 13 referred to as t(2;13) [1, 3, 6, 8, 12]. The location and size of the primary tumor and associated metastases determine the presenting symptoms and signs of the disease [8]. This cancer does not secrete any hormones or hormone-like peptides and is therefore not associated with any paraneoplastic syndromes.
The tumor is staged based on TNM classification using TNM and Intergroup Rhabdomyosarcoma Studies (IRS) using tumor (T), nodes (N) and metastases (M) to classify the RMS by stages [13]. In stage I, the tumor is localized without metastasis and usually involves the orbit, head, neck and genitourinary tract. Stage II tumor is less than 5 cm with no metastasis to lymph nodes or other distant sites. In stage III, the tumor is usually larger than 5 cm or smaller than 5 cm with spread to surrounding lymph nodes but no distant locations [14]. Lastly, stage IV involves metastases to other areas such as the bone marrow, liver, lung or bones. The lung is the most frequent site of metastasis followed by bone, bone marrow and lymph nodes. Visceral organ metastases are rare in newly diagnosed patients. The percentage of patients with histologically proven lymphatic metastases was highest for the prostate (41%), paratesticular sites (26%) and genitourinary sites overall (24%), whereas the percentage of patients with nodal metastases from extremity lesions was 12%. The primary tumor mean diameter was significantly larger in the group with nodal metastases [15]. Sentinel lymph node biopsy is a reliable staging technique and yield positive results. It was positive most commonly associated with alveolar RMS, followed by Ewing sarcoma, myxoid liposarcoma, epitheloid sarcoma and positive in one patient with embryonal RMS out of 29 patients’ studied [16].
The treatment usually consists of surgical excision with a chemotherapy regimen consisting of vincristine, dactinomycin (actinomycin-D) and cyclophosphamide. Other agents that have been used include ifosfamide, etoposide and doxorubicin [5, 8]. It is usually given once a week for the first few months, with the total length of treatment lasting from 6 months to a year. Radiation therapy is administered with chemotherapy if there is incomplete resection [17, 18]. Adherence to immunization schedule (diphtheria, pertussis, tetanus, measles, mumps, rubella and oral polio vaccine) has been shown to have some protective effect preventing development of RMS [19]. Prenatal exposure to ionizing radiation increases the risk of RMS in offspring [20].
This case discussion is extremely important due to the rarity of spindle cell carcinoma. On a search of PubMed, there are no articles written discussing the occurrence and prognosis of the sclerosing spindle cell variant. While RMS is more common in males, this case discussion is of a 7-year-old female patient. In addition, spindle cell most commonly occurs in the orbital and para-testicular areas as opposed to the pre-auricular location seen in this patient. The spindle cell embryonal subtype is not known to metastasize to lymph nodes [3, 17, 20, 21]. According to this case, surgery, radiation and chemotherapy have resulted in a positive prognosis. A wider discussion needs to be focused on this variant of RMS to help determine the prognosis and treatment approach.
Conclusions
Any time a growing mass in the head and neck location is encountered, an aggressive approach must be taken and RMS must be on our differential diagnosis. Early diagnosis and a multidisciplinary approach to the management of patients with RMS give the best prognostic outlook. The tumor is sensitive to radiation and chemotherapy, after the early excision of the mass along with removal of involved nodes. Our case is unique with a diagnosis of spindle cell RMS discovered in the preauricular location in a female patient. Our objectives in reporting this unusual case is to increase awareness among practitioners and participate in formation of the database. Further research needs to be done to understand the pathology and spread of this subtype of RMS.
Financial Support
No grants or financial support received.
Conflict of Interest
Authors have no conflict of interest.
| References | ▴Top |
- Dziuba I, Kurzawa P, Dopierala M, Larque AB, Januszkiewicz-Lewandowska D. Rhabdomyosarcoma in children - current pathologic and molecular classification. Pol J Pathol. 2018;69(1):20-32.
- Crozier E, Rihani J, Koral K, Cope-Yokoyama S, Rakheja D, Ulualp SO. Embryonal rhabdomyosarcoma of the auricle in a child. Pediatr Int. 2012;54(6):945-947.
doi - Wolden SL, Alektiar KM. Sarcomas across the age spectrum. Semin Radiat Oncol. 2010;20(1):45-51.
doi - Kumar A, Singh M, Sharma MC, Bakshi S, Sharma BS. Pediatric sclerosing rhabdomyosarcomas: a review. ISRN Oncol. 2014;2014:640195.
- Rahman HA, Sedky M, Mohsen I, Taha H, Loaye I, Zaghloul MS, Wakeel ME, et al. Outcome of pediatric parameningeal rhabdomyosarcoma. The Children Cancer Hospital, Egypt, experience. J Egypt Natl Canc Inst. 2013;25(2):79-86.
doi - Hayes-Jordan A, Andrassy R. Rhabdomyosarcoma in children. Curr Opin Pediatr. 2009;21(3):373-378.
doi - Shrutha SP, Vinit GB. Rhabdomyosarcoma in a pediatric patient: A rare case report. Contemp Clin Dent. 2015;6(1):113-115.
doi - Arndt CA, Rose PS, Folpe AL, Laack NN. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin Proc. 2012;87(5):475-487.
doi - Owosho AABCD, Huang SCM, Chen SM, Kashikar SD, Estilo CLD, Wolden SLM, Wexler LHM, et al. A clinicopathologic study of head and neck rhabdomyosarcomas showing FOXO1 fusion-positive alveolar and MYOD1-mutant sclerosing are associated with unfavorable outcome. Oral Oncol. 2016;61:89-97.
doi - Robinson JC, Richardson MS, Neville BW, Day TA, Chi AC. Sclerosing rhabdomyosarcoma: report of a case arising in the head and neck of an adult and review of the literature. Head Neck Pathol. 2013;7(2):193-202.
doi - Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27(20):3391-3397.
doi - Salem B, Hofherr S, Turner J, Doros L, Smpokou P. Childhood Rhabdomyosarcoma in Association With a RASopathy Clinical Phenotype and Mosaic Germline SOS1 Duplication. J Pediatr Hematol Oncol. 2016;38(8):e278-e282.
doi - Embryonal Rhabdomyosarcoma. (n.d.). Retrieved from http://surgpathcriteria.stanford.edu/srbc/rhabdomyosarcoma/embryonal.html.
- Hays DM. Rhabdomyosarcoma. Clin Orthop Relat Res. 1993;289:36-49.
- Lawrence W, Jr., Hays DM, Heyn R, Tefft M, Crist W, Beltangady M, Newton W, Jr., et al. Lymphatic metastases with childhood rhabdomyosarcoma. A report from the Intergroup Rhabdomyosarcoma Study. Cancer. 1987;60(4):910-915.
doi - Dall'Igna P, De Corti F, Alaggio R, Cecchetto G. Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg. 2014;24(6):482-487.
doi - Raney RB, Walterhouse DO, Meza JL, Andrassy RJ, Breneman JC, Crist WM, Maurer HM, et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J Clin Oncol. 2011;29(10):1312-1318.
doi - Walterhouse D, Pappo AS, Meza JL, Breneman JC, Hayes-Jordan AA, Parham D, Cripe TP, et al. Shorter duration therapy that includes vincristine (V), dactinomycin (A), and lower doses of cyclophosphamide (C) with or without radiation therapy for patients with newly diagnosed low-risk embryonal rhabdomyosarcoma (ERMS): A report from the Children's Oncology Group (COG). Journal of Clinical Oncology. 2011;29(15_suppl):9516-9516.
- Sankaran H, Danysh HE, Scheurer ME, Okcu MF, Skapek SX, Hawkins DS, Spector LG, et al. The role of childhood infections and immunizations on childhood rhabdomyosarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2016;63(9):1557-1562.
doi - Brent RL. Carcinogenic risks of prenatal ionizing radiation. Semin Fetal Neonatal Med. 2014;19(3):203-213.
doi - Dagher R, Helman L. Rhabdomyosarcoma: an overview. Oncologist. 1999;4(1):34-44.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.