

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 6, Number 12, December 2015, pages 547-553
Contraction of a Maternal Fragile X Mental Retardation 1 Premutation Allele
Patricia Mirandaa, Poonnada Jiraanonta, b, Liane J. Abramsc, Kirin K. Basutaa, Janey Youngblomd, Andrea Schneidere, f, Randi J. Hagermane, f, Flora Tassonea, e, g
aDepartment of Biochemistry and Molecular Medicine, School of Medicine, University of California Davis, Davis, CA, USA
bResearch Center for Neuroscience, Institute of Molecular Biosciences, Mahidol University, Nakornpathom, Thailand
cUniversity of California, San Francisco, San Francisco, CA, USA
dDepartment of Biology, California State University Stanislaus, Turlock, CA, USA
eMIND Institute, University of California Davis Medical Center, Sacramento, CA, USA
fDepartment of Pediatrics, School of Medicine, University of California Davis, Davis, CA, USA
gCorresponding Author: Flora Tassone, Department of Biochemistry and Molecular Medicine, MIND Institute, 2805 50th Street, Sacramento, CA 95817, USA
Manuscript accepted for publication September 17, 2015
Short title: Instability of the FMR1 Premutation Allele
doi: http://dx.doi.org/10.14740/jmc2301w
| Abstract | ▴Top |
Fragile X syndrome (FXS) is mainly caused by an expansion of a cytosine-guanine-guanine (CGG) trinucleotide repeat (greater than 200) in the 5’ untranslated region of the fragile X mental retardation 1 (FMR1) gene resulting in gene silencing, methylation, and absence of FMRP. FXS involves a wide spectrum of phenotypes including intellectual disabilities, autism spectrum disorder (ASD), maladaptive behaviors or symptoms, social anxiety, mood disorders and physical features. Carriers of premutation alleles (55 - 200 CGG repeats) can manifest with neurodevelopmental phenotypes and are at risk for the neurodegenerative fragile X-associated tremor/ataxia syndrome (FXTAS) and about 20% of women with a premutation develop fragile X-associated primary ovarian insufficiency (FXPOI). These phenotypes are believed to be caused by a distinct molecular mechanism involving increased FMR1 mRNA production and toxicity. Premutation alleles are unstable and depending on the CGG length and the presence of AGG interruptions have the potential to expand to larger premutation or full mutation alleles during transmission from mothers to their children. The contraction of a CGG repeat allele, although rare, has been observed. Here, we report the case of a three-generation family, who was identified as part of a pilot study of newborn screening for FXS. Specifically, the maternal grandmother carried a premutation allele (109 CGG repeats), which contracted to an intermediate size allele (52 CGG repeats) in her son, the father of a newborn girl (proband). This allele expanded to a premutation allele of 59 CGG repeats in the proband newborn. A premutation allele of 61 CGG repeats was detected in the proband’s sister who demonstrated social phobia but normal cognition. The proband newborn showed some fragile X associated physical stigmata but normal cognition. Intriguingly, all FMR1 CGG expanded allele showed no AGG interruptions. This case shows a remarkable instability of premutation alleles throughout the three generations likely arising from different mechanisms.
Keywords: Premutation; Fragile X; FMR1; CGG contraction; Transmission
| Introduction | ▴Top |
Fragile X syndrome (FXS) is the most common inherited cause of intellectual disability and the most common single gene cause of autism. This condition is caused by a cytosine-guanine-guanine (CGG) trinucleotide expansion in the 5’ untranslated region of the fragile X mental retardation 1 (FMR1) gene, which encodes for the fragile X mental retardation protein (FMRP). This expansion leads to inhibition of gene transcription, gene methylation, and consequent absence of FMRP [1].
Variability in the CGG repeat numbers defines the four-allele categories. Individuals with CGG repeat numbers greater than 200 have the full mutation or FXS, which results in decreased levels or absent FMRP. Some of the hallmark physical characteristics of FXS in males include large and prominent ears, long narrow face, hyper-extensible joints, strabismus, high-arched palate, and macro-orchidism. Social, behavioral, and emotional characteristics include anxiety, attention deficit hyperactivity disorder (ADHD), hypersensitivity to sensory stimuli, hand flapping, and poor eye contact. While males with FXS usually have moderate to severe intellectual disability (ID), females with a full mutation, display phenotypic involvement ranging from normal cognitive function to ID, depending on X inactivation and resulting levels of FMRP [1, 2]. Affected females with FXS have social anxiety, learning and behavioral disorders, and extreme shyness [3-5].
Individuals with 55 - 200 CGG repeat alleles have the FMR1 premutation and produce high FMR1 mRNA levels, leading to RNA toxicity, and FMRP expression levels vary from normal to moderately lower FMRP levels [6-8]. Until recently, premutation carriers were considered to be clinically unaffected, but a range of clinical phenotypes have now been well described [9]. Individuals with the premutation can present with physical, cognitive, and behavioral features that are also seen in individuals with FXS including ADHD, autism spectrum disorder (ASD), and anxiety [10-12]. About 20% of women with the fragile X premutation experience fragile X-associated primary ovarian insufficiency (FXPOI), or menopause before age 40, compared to 1% in the general population [13]. Fragile X-associated tremor ataxia syndrome (FXTAS), characterized by tremor, ataxia, peripheral neuropathy, and loss of memory and cognitive function, is observed in over 40% of male premutation carriers and about 8-16% of female premutation carriers [14-16].
Individuals with the gray zone or intermediate allele have 45 - 54 CGG repeats. Although there is limited information on gray zone allele carriers, a number of studies have shown clinical involvement similar to premutation carriers in these individuals. Recent studies and case reports have shown an association between gray zone alleles and an increased risk for conditions such as parkinsonism, FXTAS, and FXPOI [17-20].
FMR1 alleles are inherited in an X-linked manner. FMR1 alleles with CGG repeats in the normal range are typically stably transmitted, while gray zone and premutation alleles are unstable and can expand from generation to generation [21, 22]. A full mutation arises from the expansion of a maternal premutation allele. The risk of a maternal premutation allele expanding to a full mutation during transmission is influenced by the presence of AGG interruptions within the alleles [21, 23], which can also affect the magnitude of expansion, particularly in the intermediate and premutation range [21, 22].
A man with a premutation or full mutation allele passes on a premutation allele to all his daughters and the increase in size is generally small [24]. Cases of contractions of maternal premutation alleles have been reported, but are rare [25-27].
Here, we report on a family in which a maternal premutation allele contracts to about half the number of repeats (Fig. 1) in her son, down to an intermediate allele, which expanded during transmission to a premutation allele in his two daughters.
 Click for large image | Figure 1. Pedigree of a family with an atypical transmission of the FMR1 allele over three generations. II-2 is the individual with a premutation allele of 109 CGG repeats which contracted in the following generation. The proband with a premutation allele identified through the NBS pilot project is indicated by an arrow. A dot within a square or a circle indicates that the corresponding individual has a premutation or intermediate allele. |
| Case Report | ▴Top |
Father (III-2) is a 53-year-old Caucasian male identified as carrier of an FMR1 intermediate allele of 52 CGG repeats, following positive FMR1 newborn screening results on his daughter, IV-4. III-2 was clinically evaluated at 52 years old at the UC Davis MIND Institute. His medical history was remarkable for sleep apnea, snoring, hypertension, carpal tunnel, and numbness in his left middle finger. He is taking three medications to control his hypertension. His birth and development history were normal except for learning problems in school related to ADHD symptoms.
Upon physical examination, he presented with height of 178.2 cm (50 - 75th percentile), weight of 101.1 kg (75 - 95th percentile), and an occipital frontal circumference (OFC) of 58 cm (97th percentile) and his vitals demonstrated significant hypertension 166/110. His exam was negative for features such as a long, narrow face, prominent ears, ear cupping, epicanthal folds, strabismus, ptosis, high palate, macro-orchidism, hyper-extensible finger joints and prominent jaw. Completely flat feet were observed. His cardiac and neurology evaluations were unremarkable.
Wechsler adult intelligence scale III (WAIS-III) test showed average verbal (verbal IQ: 92), performance (performance IQ: 107), and full-scale IQ scores (FSIQ: 99). His WAIS working memory score (WM: 92) and processing speed (PS: 103) fell in the average range. Based on Wechsler memory scale III (WMS-III) test, his auditory (auditory immediate: 94; auditory delayed: 105), visual (visual immediate: 109; visual delayed: 103), and general memory scores (general memory: 101) also fell in the average range [28-30]. On the behavioral dyscontrol scale, a measure of executive functioning on frontal lobe control [31], the participant scored an average 21 points. His Purdue Pegboard test [32] did not show any fine motor problems.
III-2 completed a survey for another study administered through the UC Davis MIND Institute approximately 2 years after his clinical evaluation. In the survey, he reported symptoms of numbing in his hands and fingers and memory loss. He also reported taking a supplement called LifeVantage Protandim to prevent and/or treat any symptoms of FXTAS.
III-2 has five children, three males and two females. All of his sons had CGG repeats in the normal range. The proband (IV-4), as mentioned above, was found to have 37 and 59 CGG repeats through UC Davis Fragile X Newborn Screening Pilot Study [33, 34]. Sister of the proband (IV-2) was found to have 37 and 61 CGG repeats. Grandmother of the proband, II-2, was tested and was found to have 14 and 109 repeats; thus during transmission, the maternal premutation allele contracted to 52 CGG repeats in III-2. None of the premutation alleles in II-2, IV-4, and IV-2 had AGG interruptions; therefore, all contained uninterrupted CGG stretches.
The proband, IV-4, was the product of an uncomplicated term pregnancy with a birth weight of 4 kg. There were no recognizable malformations at birth. Regarding her development, she was appropriately meeting her developmental milestones: she was sitting unassisted at 6 months, crawling around 7 - 8 months, walking at 12 months, speaking single words at 9 months, and using phrases at 24 months. At 2 years and 6 months of age, her mother reports the following behaviors: chewing on things, tantrums, shyness, sleep disturbances, anxiety (new situations only), occasional moodiness/irritability, and taking off clothing without being asked, although all of these symptoms are within the normal range for her age.
During her most recent physical examination at age of 2 years and 6 months, she was noted to have the following characteristics: prominent ears, ear cupping and completely flat feet/minimal arch. Her OFC was 51 cm (98th percentile) and height was 89.4 cm (50th percentile). Both cardiac and neurology examinations were normal.
IV-4’s scores from her Mullen scales of early learning, which assesses cognitive functioning, and Vineland-II, which measures personal and social skills, are summarized in Table 1. The ADOS, a semistructured observational instrument used to evaluate for ASD, yielded a “typical” (non-autism) diagnosis.
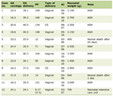 Click to view | Table 1. IV-4 - Developmental Assessment Results |
Sister of the proband, IV-2, underwent FMR1 DNA testing after her younger sister IV-4 was identified to be a premutation carrier from NBS. IV-2 was determined to also be a premutation carrier, with two alleles of 37 and 61 CGG repeats, respectively. Her birth history and developmental milestones were normal and at 11 years on exam, she did not have features of FXS. However, she has a history of significant anxiety, school phobia, social phobia and depression with previous suicidal ideation. The Kiddie-Schedule for affective disorders and schizophrenia (K-SADS) [35] interview provided the following diagnoses for IV-2: social phobia (generalized performance situation, public bathrooms), psychotic d/o NOS (persistent auditory hallucinations, some visual, but no impairment in social functioning), children’s global assessment scale (CGAS): 70 (some mild symptoms). Results from the Wechsler intelligence scale for children-IV (WISC-IV) [36] showed generally results in the average range: a verbal comprehension index of 100, perceptual reasoning index of 110, working memory index of 102, processing speed index of 83 (low average), and a full-scale IQ of 101. She was treated with counseling, biofeedback and sertraline with significant improvement in her symptoms.
Cascade testing of the family identified III-2’s mother (II-2) as a premutation carrier with a normal allele of 14 CGG repeats and a premutation allele of 109 CGG repeats, which contracted to 52 CGG repeats during transmission. She was clinically evaluated at the UC Davis MIND Institute at 68 years old. II-2 reported no known congenital malformations and complications during her birth. She reportedly had a normal developmental history. Her medical history includes gall bladder surgery at 63 years old, ablation for arrhythmia at 65 years old, and a hysterectomy at 32 years old. She also has hypertension.
Upon physical examination, II-2 had a long and/or narrow face and strabismus. She does not have flat feet. Her OFC is 52.5 cm (third - 10th percentile) and her height is 159 cm (25th - 50th percentile). Her cardiac evaluation noted an irregular heartbeat, while her neurological exam was normal except for a mild tremor on the left side with finger to nose testing. Fragile X DNA molecular testing was performed on all subjects using both PCR and Southern blot analysis [37, 38]. Results are shown in Figure 2.
 Click for large image | Figure 2. CGG size instability. Electrophoregrams (CE) illustrate the four cases including the grandmother with a premutation (II-2), the father carrier of an intermediate allele (III-2), and the two premutation daughters (IV-2 and IV-4). The electrophoregrams visualize the PCR amplicons obtained by triplet-primer PCR (TR-PCR) method as described in Sadic-Filipovik et al, 2010. The x-axis represents the size of the alleles in base pairs. The y-axis represents the fluorescence intensity. Southern Blot data are presented on the right side of the CE and show the presence of an expanded alleles in all four cases (lane 1 in a, b, c and d). M: 1 kb size marker. C: negative control, female normal. Southern blot analysis was conducted as described in Tassone et al [37]. |
| Discussion | ▴Top |
The mechanism of FMR1 instability remains to be clarified. Recent studies propose that genetic and environmental factors may influence the process of expansions differently from those leading to repeat contractions [24, 39], though their outcomes show the same underlying process. The bulk of evidence suggests that expansion can occur during transcription or open chromatin configuration involving DNA repair independently from genomic replication. Specifically, the CGG repeats can form a number of different structures, like hairpins/stem loops, during DNA replication and somehow be responsible for the expansion. A variety of proteins involved in DNA repair and recombination are also likely to be involved in the process of repeat expansion [40].
Current evidence indicates that large repeat expansion may occur via base excision repair (BER) or failure of 5’ flap. During removal of oxidized DNA bases by BER, the loop formation originates instead from repair of single-strand breaks (nicking); consequently, gap-filling synthesis to restore the correct bases and ligation to recombine the broken end, complete the process. In BER, gap-filling synthesis can happen either via a long patch or a short patch mechanism [41]. The long patch BER has been suggested as the crucial pathway for DNA repair and large repeat expansions, which involve DNA repair polymerase β (Polβ), Polδ, and Polε [42]. CGG repeat could also arise by a hairpin structure formation on the displaced strand within 5’ flap and ligation of this hairpin to the next Okazaki fragment repeated during replication cycles causing large expansion [40, 43].
The molecular mechanisms responsible for the rare occurrence of CGG repeat contractions in the FMR1 gene are complex and still need to be defined. DNA polymerase slippage or slipped strand mis-pairing (SSM) may be leading to the reduction of the CGG length. The contraction process generally occurs in GC-rich repetitive sequences possibly by transient detachment of newly synthesized strand and mis-pairing during DNA replication because of the repetitive nature of the CGG stretch [44, 45]. It was demonstrated that CpG methylation and SSM may facilitate the formation of deletion events. Consequently, CpG-rich region may be prone to rearrangements, which results in deletion that can extend into the promoter sequence preceding CGG repeat stretch [46]. Deletions may also result from intramolecular recombination relating to direct repeats or double-strand break repair between the promotor region and the CGG repeat tract. In contrast, SSM leads to greater or lesser number of CGG repeats as a consequence of misalignment of either the newly synthesized or template strand [25]. Moreover, CGG deletion observed in spermatogonia of males with FXS implies that large deletion could occur by multiple slippage events that arise while the DNA polymerase copies the trinucleotide tracts. If the DNA polymerase cannot pass through a repeat tract, it will induce the expression of nucleotide excision repair machinery and translation polymerase to bypass this block and restart replication. This single-strand repeat loop on the template strand would thus be deleted in the next round of replication [47-49].
The contraction of FMR1 alleles when transmitted from parent-to-child has been previously reported in the literature with the majority of the deletions including the CGG repeat occurring during transmission of a maternal premutation allele to the offspring [50]. Coffee et al [50] reported three unrelated females who had normal size FMR1 alleles despite their mothers being premutation carriers. The contraction in repeat size from mother to daughter was 95 to 35 CGG repeats in one family, 145 to 43 CGG repeats in another family, and 82 to 33 CGG repeats in the third family [26]. Vits et al [27] reported a contraction from 110 CGG repeats in the mother to 44 CGG repeats in her daughter [27]. In Alfaro et al [51], a mother’s premutation (93 CGG repeats) expanded to > 200 CGG repeats in one male and contracted to 54 CGG repeats in a female twin [51]. Tabolacci et al [25] described the case of reversion from a maternal premutation allele of about 190 CGG repeat to a normal size allele of 43 CGG repeats in the male child.
In this study, we report a family identified through an NBS pilot project in which both CGG repeat contraction and expansion are distributed through the instability of FMR1 alleles with no AGG interruptions. The grandmother with 109 CGG repeats transmitted an intermediate size of 52 CGG repeats to her son resulting from a reduction of 57 CGG repeats. The intermediate paternal allele expanded during transmission to premutation alleles of 61 and 59 CGG repeats in the two daughters (Fig. 1).
Increasing maternal CGG allele size has been considered the most important risk of having off spring with the full mutation [52] since detection of the FMR1 gene in 1991. Stability of alleles during transmission is influenced by the total length of the CGG allele. However, the role of AGG interruptions has been recently established and a risk model for expansion to a full mutation during transmission has been proposed [21, 22]. These studies demonstrated that maternal alleles with no AGGs had a greater frequency and magnitude of instability on transmission than those with one or two AGG interruptions. In addition, they indicated that a model taking in consideration total CGG length, number of AGG interruptions, and maternal age is recommended for calculating the predictive risk of expansion of a premutation allele to a full mutation during maternal transmission [22].
In the family reported here, the premutation allele of 59 CGG repeats identified in the newborn and of 61 CGG repeat identified in her sibling contain no AGG interruptions and have an approximate risk of expansion to a full mutation up to 23% depending on maternal age [22].
In conclusion, this study reports on a three-generation family in which both a contraction from a premutation to an intermediate unstable allele and expansion back to premutation alleles with no AGG interruptions, are observed.
| References | ▴Top |
- Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1(6):397-400.
doi pubmed - Hagerman RJ, Hagerman PJ. Fragile X syndrome: Diagnosis, treatment, and research. Baltimore, Maryland: The Johns Hopkins University Press; 2002.
doi - Roberts JE, Bailey DB, Jr., Mankowski J, Ford A, Sideris J, Weisenfeld LA, Heath TM, et al. Mood and anxiety disorders in females with the FMR1 premutation. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(1):130-139.
doi pubmed - Riddle JE, Cheema A, Sobesky WE, Gardner SC, Taylor AK, Pennington BF, Hagerman RJ. Phenotypic involvement in females with the FMR1 gene mutation. Am J Ment Retard. 1998;102(6):590-601.
doi - Hunter JE, Allen EG, Abramowitz A, Rusin M, Leslie M, Novak G, Hamilton D, et al. Investigation of phenotypes associated with mood and anxiety among male and female fragile X premutation carriers. Behav Genet. 2008;38(5):493-502.
doi pubmed - Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000;66(1):6-15.
doi pubmed - Kennerson ML, Zhu D, Gardner RJ, Storey E, Merory J, Robertson SP, Nicholson GA. Dominant intermediate Charcot-Marie-Tooth neuropathy maps to chromosome 19p12-p13.2. Am J Hum Genet. 2001;69(4):883-888.
doi pubmed - Primerano B, Tassone F, Hagerman RJ, Hagerman P, Amaldi F, Bagni C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA. 2002;8(12):1482-1488.
pubmed - Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013;12(8):786-798.
doi - Aziz M, Stathopulu E, Callias M, Taylor C, Turk J, Oostra B, Willemsen R, et al. Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet B Neuropsychiatr Genet. 2003;121B(1):119-127.
doi pubmed - Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27(2 Suppl):S137-144.
doi pubmed - Chonchaiya W, Au J, Schneider A, Hessl D, Harris SW, Laird M, Mu Y, et al. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum Genet. 2012;131(4):581-589.
doi pubmed - Sherman SL. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97(3):189-194.
doi - Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57(1):127-130.
doi pubmed - Jacquemont S, Hagerman RJ, Leehey MA, Hall DA, Levine RA, Brunberg JA, Zhang L, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291(4):460-469.
doi pubmed - Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, Pan R, Bronsky HE, et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146A(8):1009-1016.
doi pubmed - Bretherick KL, Fluker MR, Robinson WP. FMR1 repeat sizes in the gray zone and high end of the normal range are associated with premature ovarian failure. Hum Genet. 2005;117(4):376-382.
doi pubmed - Hall DA, Berry-Kravis E, Zhang W, Tassone F, Spector E, Zerbe G, Hagerman PJ, et al. FMR1 gray-zone alleles: association with Parkinson's disease in women? Mov Disord. 2011;26(10):1900-1906.
doi pubmed - Liu Y, Winarni TI, Zhang L, Tassone F, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome (FXTAS) in grey zone carriers. Clin Genet. 2013;84(1):74-77.
doi pubmed - Loesch DZ, Tassone F, Lo J, Slater HR, Hills LV, Bui MQ, Silburn PA, et al. New evidence for, and challenges in, linking small CGG repeat expansion FMR1 alleles with Parkinson's disease. Clin Genet. 2013;84(4):382-385.
doi pubmed - Nolin SL, Sah S, Glicksman A, Sherman SL, Allen E, Berry-Kravis E, Tassone F, et al. Fragile X AGG analysis provides new risk predictions for 45-69 repeat alleles. Am J Med Genet A. 2013;161A(4):771-778.
doi pubmed - Yrigollen CM, Martorell L, Durbin-Johnson B, Naudo M, Genoves J, Murgia A, Polli R, et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J Neurodev Disord. 2014;6(1):24.
doi pubmed - Yrigollen CM, Durbin-Johnson B, Gane L, Nelson DL, Hagerman R, Hagerman PJ, Tassone F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med. 2012;14(8):729-736.
doi pubmed - Nolin SL, Glicksman A, Ersalesi N, Dobkin C, Brown WT, Cao R, Blatt E, et al. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet Med. 2015;17(5):358-364.
doi pubmed - Tabolacci E, Pomponi MG, Pietrobono R, Chiurazzi P, Neri G. A unique case of reversion to normal size of a maternal premutation FMR1 allele in a normal boy. Eur J Hum Genet. 2008;16(2):209-214.
doi pubmed - Brown WT, Houck GE, Jr., Ding X, Zhong N, Nolin S, Glicksman A, Dobkin C, et al. Reverse mutations in the fragile X syndrome. Am J Med Genet. 1996;64(2):287-292.
doi - Vits L, De Boulle K, Reyniers E, Handig I, Darby JK, Oostra B, Willems PJ. Apparent regression of the CGG repeat in FMR1 to an allele of normal size. Hum Genet. 1994;94(5):523-526.
doi pubmed - Wechsler D. Wechsler Adult Intelligence Scale. third edition ed. London: The Psychological Corporation Limited; 1997.
- Wechsler D. Wechsler memory scale third edition ed. New York Psychological Corporation; 1997.
- Wechsler D. WAIS-III/WMS-III: Technical manual (updated). San Antonio, TX: The Psychological Corporation; 2002.
- Grigsby JK. The behavioral dyscontrol scale: manual 2nd ed. Denver Ward, CO: BDS.; 1996.
- Tiffin J. Purdue Pegboard: Examiner manual. . Chicago: Science Research Associates; 1968.
- Tassone F, Iong KP, Tong TH, Lo J, Gane LW, Berry-Kravis E, Nguyen D, et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012;4(12):100.
doi pubmed - Sorensen PL, Gane LW, Yarborough M, Hagerman RJ, Tassone F. Newborn screening and cascade testing for FMR1 mutations. Am J Med Genet A. 2013;161A(1):59-69.
doi pubmed - Kaufman J, Birmaher B, Brent D, Rao U, Flynn C, Moreci P, Williamson D, et al. Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL): initial reliability and validity data. J Am Acad Child Adolesc Psychiatry. 1997;36(7):980-988.
doi pubmed - Wechsler D. Wechsler Intelligence Scale for children, 4th ed. (WISC-IV). 4th edition ed. San Antonio, TX: The Psychological Corporation; 2003.
- Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10(1):43-49.
doi pubmed - Filipovic-Sadic S, Sah S, Chen L, Krosting J, Sekinger E, Zhang W, Hagerman PJ, et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem. 2010;56(3):399-408.
doi - Lokanga RA, Zhao XN, Usdin K. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum Mutat. 2014;35(1):129-136.
doi pubmed - Zhao XN, Usdin K. The Repeat Expansion Diseases: The dark side of DNA repair. DNA Repair (Amst). 2015;32:96-105.
doi pubmed - Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003;531(1-2):37-80.
doi pubmed - Satter EK, Graham BS, Steger JW. Response of cutaneous Rosai-Dorfman disease to topical and intralesional steroids. Br J Dermatol. 2003;149(3):672-674.
doi pubmed - Kim JC, Mirkin SM. The balancing act of DNA repeat expansions. Curr Opin Genet Dev. 2013;23(3):280-288.
doi pubmed - Chiurazzi P, Kozak L, Neri G. Unstable triplets and their mutational mechanism: size reduction of the CGG repeat vs. germline mosaicism in the fragile X syndrome. Am J Med Genet. 1994;51(4):517-521.
doi pubmed - Garcia Arocena D, de Diego Y, Oostra BA, Willemsen R, Mirta Rodriguez M. A fragile X case with an amplification/deletion mosaic pattern. Hum Genet. 2000;106(3):366-369.
doi pubmed - Nichol Edamura K, Pearson CE. DNA methylation and replication: implications for the "deletion hotspot" region of FMR1. Hum Genet. 2005;118(2):301-304.
doi pubmed - Reyniers E, Vits L, De Boulle K, Van Roy B, Van Velzen D, de Graaff E, Verkerk AJ, et al. The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat Genet. 1993;4(2):143-146.
doi pubmed - Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73(1):134-154.
doi pubmed - McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11(11):786-799.
doi pubmed - Coffee B, Ikeda M, Budimirovic DB, Hjelm LN, Kaufmann WE, Warren ST. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: a case report and review of the literature. Am J Med Genet A. 2008;146A(10):1358-1367.
doi pubmed - Alfaro MP, Cohen M, Vnencak-Jones CL. Maternal FMR1 premutation allele expansion and contraction in fraternal twins. Am J Med Genet A. 2013;161A(10):2620-2625.
pubmed - Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67(6):1047-1058.
doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.