

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 4, Number 8, August 2013, pages 526-529
Primary Diffuse Leptomeningeal Gliomatosis Coincident With a Melanocytic Disorder
Julan Vimalenthiran Amalaseelana, d, David McDowellb, Michael Jerome Mckaya, b, c
aNorth Coast Cancer Institute, 70-72 Hunter Street, Lismore, NSW 2480, Australia
bDepartment of Neurosurgery, The Canberra Hospital, Yamba Drive, Garran, ACT 2605, Australia
cFaculty of Medicine, The University of Sydney, Australia
dCorresponding author: Julan Vimalenthiran Amalaseelan, North Coast Cancer Institute, 70-72 Hunter Street, Lismore, NSW 2480, Australia
Manuscript accepted for publication June 6, 2013
Short title: Primary Diffuse Leptomeningeal Gliomatosis
doi: https://doi.org/10.4021/jmc1361w
| Abstract | ▴Top |
Primary diffuse leptomeningeal gliomatosis (PDLG) is a rare neoplasm of the leptomeninges associated with limited survival time. Around 60 cases of PDLG have been reported in the literature and most of them were diagnosed at post mortem examination. We describe the longest survival in a PDLG case, a 24 year old man who survived for 30 months with an aggressive treatment approach that included chemoradiation and repeated surgical excisions of tumor. Our patient also had multiple dysplastic nevi and dysmorphic facial features raising the suspicion of an underlying genetic disorder.
Keywords: Primary diffused leptomeningeal gliomatosis; Dysplastic nevus syndrome
| Introduction | ▴Top |
Primary diffuse leptomeningeal gliomatosis (PDLG) is a rare malignant condition characterized by infiltration, often widespread, of the leptomeninges with glioma cells, without evidence of a primary tumor within the brain or spinal cord parenchyma [1]. The survival of patients with PDLG is limited and the longest survival reported in the literature was 24 months [2]. The association of gliomas and melanocytic disorders are rarely described [3-5].
We describe a patient with PDLG who survived for 30 months with combined modality treatments including surgery, radiotherapy and chemotherapy. This is the longest survival documented for a patient with PDLG. He was also found to have multiple melanocytic skin lesions suggestive of dysplastic nevus syndrome, and two first degree relatives with melanocytic disorders.
| Case Report | ▴Top |
A 24-year-old overweight Caucasian male presented with progressive frontal headache of 6 months duration. The headache was worse during early mornings and was exacerbated by straining. A contrast CT of the brain performed a few months prior to this was reported as normal. He was referred to a neurology outpatient clinic for further evaluation. In the interim, he was admitted to an emergency unit with severe headache and unsteady gait. On examination, his Glasgow Coma Scale was 15/15 and blood pressure was 110/60 mm Hg with a pulse rate of 80/min. Fundoscopic examination revealed bilateral florid papilloedema. No focal neurological signs were found. He had frontal bossing and multiple nevi on his skin (Fig. 1).
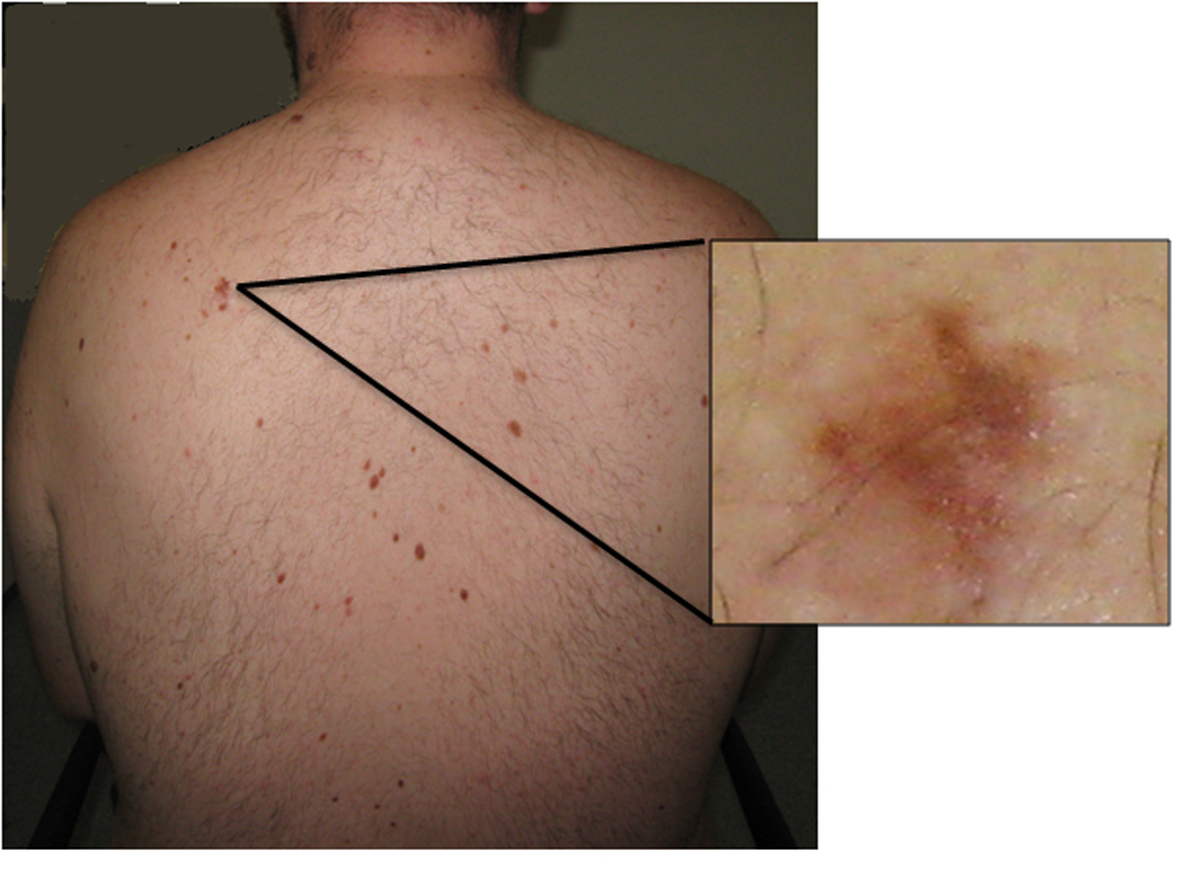 Click for large image | Figure 1. Photograph showing the back of the patient. Multiple pigmented dysmorphic nevi are evident. Inset: higher power view of a dysmorphic nevus. |
The CT of his brain on admission was reported as normal. A lumbar puncture was performed with CSF manometry. An opening CSF pressure of > 50 cm H2O (normal range: 7 - 18 cm) was measured. He noticed immediate relief of headache after the lumbar puncture. CSF analysis showed elevated leucocyte counts of 30/mm3 (normal range: 0 - 5/mm3) with polymorphs of 28/mm3 (normal value: 0/mm3). The CSF protein level was elevated to 1836 mg/L (normal range: 150 - 600 mg/L). No bacterial growth was detected. Cytological examination did not show any malignant cells.
He underwent an MRI brain to rule out venous sinus thrombosis. This showed a large lesion adjacent to his left upper cerebellum, possibly extending along the subarachnoid space both anteriorly and posteriorly (Fig. 2). No brain parenchymal space occupying lesion was identified. He underwent retrosigmoid craniotomy with resection of a lesion “in the vicinity of the left cerebello-pontine angle” at surgery. The lesion was not parenchymal and appeared to lie in the subarachnoid space. The histopathology described loose sheets of malignant cells with abundant eosinophilic cytoplasm. The tumor also showed perivascular pseudorosettes, necrosis and vascular proliferation. Immunostaining showed positive GFAP, vimentin and AE 1/3 staining. A histopathological diagnosis of glioblastoma multiforme (GBM), WHO grade 4 was made. The tumor fit the criteria for primary diffuse leptomeningeal gliomatosis (PDLG).
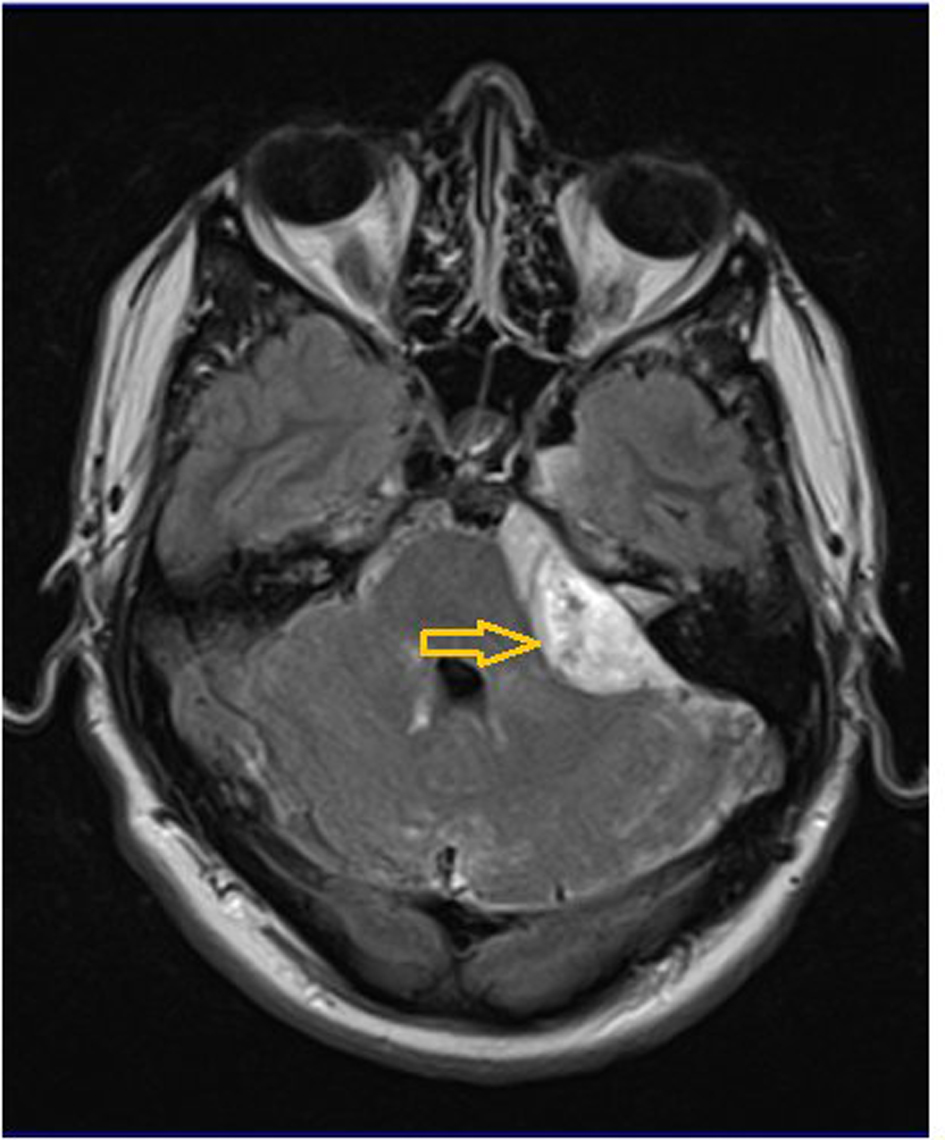 Click for large image | Figure 2. T1-weighted MRI with gadolinium contrast, axial view. There is an enhancing lesion adjacent to and compressing the left cerebellum and extending along the subarachnoid space both anteriorly and posteriorly (arrow). |
He was treated with concurrent chemoradiation with temozolomide (75 mg/m2 per day, 7 days per week from first to last day of radiotherapy). He received 54 Gy in 30 fractions to the posterior cranial fossa over 6 weeks. He then had adjuvant temozolomide every four weeks for 6 months. He remained asymptomatic during this period.
A surveillance MRI done 12 months after his radiotherapy revealed new asymptomatic multifocal leptomeningeal enhancement throughout his spinal canal. Intradural, extramedullary nodules were seen at the T9 and T10 levels. T9-10 laminectomy and biopsy confirmed GBM of the same histology as previously. He also had a parenchymal recurrence in the left frontal lobe which was surgically excised.
He had progressive intracranial and spinal disease and died 30 months after his initial diagnosis. No post-mortem was performed.
| Discussion | ▴Top |
PDLG is a rare, uniformly fatal neoplastic condition [6]. It is characterized by widespread leptomeningeal infiltration with glioma cells, without evidence of a primary tumor within the brain or spinal parenchyma [1]. It is thought to arise from heterotopic glial cells in the subarachnoid meningeal space. Heterotopic glial nests have been noted in the subarachnoid space in about 1% of unselected autopsies [7]. The incidence of PDLG is substantially higher in patients with congenital anomalies, particularly those of the central nervous system [8]. Around 60 cases of PDLG have been reported in the literature to date. Most of them were diagnosed at postmortem examination [9].
Hansen et al., described a 53-year-old patient with PDLG who survived for 24 months with combined chemoradiation with temozolomide. This was the longest reported survival in PDLG [2]. The review analyzed 14 previously reported cases of PDLG. Patients who received chemoradiation with temozolomide showed a median survival of 18 months. The median survival of all 15 patients was 8 months (range 1 - 24 months). Our patient’s survival time of 30 months is longer than that of all previously reported cases.
Genetic predispositions and disorders are suspected of being involved in approximately 1% of gliomas [10]. Rare familial syndromes known to predispose individuals to glioblastoma include neurofibromatosis type 1, Li-Fraumeni syndrome, tuberous sclerosis and Turcot's syndrome [10].
Our patient has a number of unusual physical features. He had marked frontal bossing (not shown), the significance of which is unclear but which is a feature of some genetic syndromes (e.g. Gorlin’s syndrome). He appeared to have had dysplastic nevus syndrome (DNS) for which he underwent regular surveillance. His father had invasive melanoma at the age of 45 years, and one of his siblings also had increased “moliness”. In 1993, Kaufman et al. first described a three-generation family where cutaneous malignant melanoma, cerebral astrocytoma or both developed in eight family members [3]. None of the members of this family had features of DNS. Subsequent surveys from different countries confirmed the familial congregation of cutaneous malignant melanoma-neural system tumors (MM-NST) [4, 5]. Azizi et al. studied 904 patients with cutaneous melanoma. In his series fifteen families with a total of 17 patients had at least one additional member with benign and/or malignant tumors of central nervous system. A second nervous system tumor was diagnosed in 10 patients (meningiomas (n = 9) and acostic neurilemmoma (n = 1)) Interestingly, DNS was found in the majority (20/25) of cutaneous melanoma patients who were dermatologically examined [4]. The underlying genetic defect was sought only in few MM-NST families. A gene or genes on chromosome 9p21 has been implicated in some of the cases with MM-NST syndrome [11]. No case of PDLG and melanocytic disorders has been reported so far in the literature.
The occurrence of PDLG and DNS in a young patient with a dysmorphic feature (frontal bossing) may indicate a single gene disorder. Alternatively, the brain tumor in our patient may be just a coincidence on a background of DNS. No brain tumors were reported in the other family members of our patient.
In conclusion, we report a rare brain tumor with extended survival after aggressive combined modality treatment. The young age of our patient, marked frontal bossing and the embroyological and genetic similarities between glial cells and melanocytes may indicate the existence of a genetic disorder that predisposes to gliomas and DNS in our patient. Identification of similar cases may advance the understanding of the oncogenesis of brain tumors and melanocytic disorders.
Conflict of Interest
None.
Grant Support
None.
| References | ▴Top |
- Bhatia R, Roncaroli F, Thomas P, Cheah SL, Mehta A, Glaser M, Ulbricht C. A case of primary leptomeningeal gliomatosis confined to the spinal cord. J Neurooncol. 2010;98(1):125-129.
doi pubmed - Hansen N, Wittig A, Hense J, Kastrup O, Gizewski ER, Van de Nes JA. Long survival of primary diffuse leptomeningeal gliomatosis following radiotherapy and temozolomide: case report and literature review. Eur J Med Res. 2011;16(9):415-419.
doi pubmed - Kaufman DK, Kimmel DW, Parisi JE, Michels VV. A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology. 1993;43(9):1728-1731.
doi pubmed - Azizi E, Friedman J, Pavlotsky F, Iscovich J, Bornstein A, Shafir R, Trau H, et al. Familial cutaneous malignant melanoma and tumors of the nervous system. A hereditary cancer syndrome. Cancer. 1995;76(9):1571-1578.
doi - Bahuau M, Vidaud D, Kujas M, Palangie A, Assouline B, Chaignaud-Lebreton M, Prieur M, et al. Familial aggregation of malignant melanoma/dysplastic naevi and tumours of the nervous system: an original syndrome of tumour proneness. Ann Genet. 1997;40(2):78-91.
pubmed - Riva M, Bacigaluppi S, Galli C, Citterio A, Collice M. Primary leptomeningeal gliomatosis: case report and review of the literature. Neurol Sci. 2005;26(2):129-134.
doi pubmed - Cooper IS, Kernohan JW. Heterotopic glial nests in the subarachnoid space; histopathologic characteristics, mode of origin and relation to meningeal gliomas. J Neuropathol Exp Neurol. 1951;10(1):16-29.
doi pubmed - Hirano S, Houdou S, Hasegawa M, Kamei A, Takashima S. Clinicopathologic studies on leptomeningeal glioneuronal heterotopia in congenital anomalies. Pediatr Neurol. 1992;8(6):441-444.
doi - Somja J, Boly M, Sadzot B, Moonen G, Deprez M. Primary diffuse leptomeningeal gliomatosis: an autopsy case and review of the literature. Acta Neurol Belg. 2010;110(4):325-333.
pubmed - Almairac F, Frenay M, Paquis P. [Genetic diseases and glioblastomas]. Neurochirurgie. 2010;56(6):455-458.
doi pubmed - Laud K, Marian C, Avril MF, Barrois M, Chompret A, Goldstein AM, Tucker MA, et al. Comprehensive analysis of CDKN2A (p16INK4A/p14ARF) and CDKN2B genes in 53 melanoma index cases considered to be at heightened risk of melanoma. J Med Genet. 2006;43(1):39-47.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.