

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 4, Number 11, November 2013, pages 765-768
Pulvinar Sign in Sporadic Creutzfeldt-Jakob Disease: Exploring the Diagnostic Modalities of Prion Diseases
Vijay Ravipatia, b, Lisa Mask-Bulla, Erin Halea, Bernadette Millera
aDepartment of Internal Medicine, University of Oklahoma School of Community Medicine, Tulsa, OK, USA
bCorresponding author: Vijay Ravipati, The University of Oklahoma-Tulsa, Schusterman Center 4502 E. 41st Street, Tulsa, OK 74135, USA
Manuscript accepted for publication April 26, 2013
Short title: Creutzfeldt-Jakob Disease
doi: https://doi.org/10.4021/jmc1278w
| Abstract | ▴Top |
Creutzfeldt-Jakob disease (CJD) refers to the most common transmissible spongiform encephalopathies. The distinction between a diagnosis of sporadically acquired CJD and the exogenously acquired CJD is important, and has previously been assisted by imaging modalities revealing the pulvinar sign. This case report describes a 71-year-old native-American female with rapidly deteriorating mental status, progressive personality changes, ataxia, and 50-pound weight loss. Non-contrast MRI of the brain was significant for symmetrical high diffusion signal in the bilateral pulvinar thalami. CSF studies performed by the National Prion Disease Surveillance Center showed a positive 14-3-3 level and an elevated tau conveying a positive predictive value of 72.6% for CJD. The case exemplifies commonality between sporadic CJD and variant CJD and challenges the utility of radiological findings differentiating the diseases. Our case emphasizes that the pulvinar sign may not be as specific for a diagnosing variant CJD as we once thought.
Keywords: Pulvinar Sign; Sporadic Creutzfeldt-Jakob disease; Diagnostic modality; Prion disease
| Introduction | ▴Top |
Transmissible spongiform encephalopathies (TSE) are exceedingly rare diseases (0.97 cases per 1 million) caused by the induction of a conformational change from the non-pathologic prion protein (PrPc) to a pathologic form (PrPSc), by sporadic, familial, or exogenous means [1]. Creutzfeldt-Jakob disease (CJD) refers to the most common form of TSE. The distinction between a diagnosis of sporadically acquired CJD and the exogenously acquired CJD is important, but often difficult to distinguish. Imaging modalities revealing the pulvinar sign have previously assisted diagnosis [2]. A diagnosis of probable sporadic CJD (sCJD) or suspected variant CJD (vCJD) may be made by combining a clinical presentation with detection of CSF protein markers, typical EEG, and MRI findings as defined by WHO criteria [3, 4].
| Case Report | ▴Top |
A 71-year-old American Indian female from Oklahoma presented with rapidly deteriorating mental status and a 50-pound weight loss over a 3-month period. Her family reported no family history or iatrogenic exposure to CJD. They also identified progressive personality changes, worsening appetite, visual hallucinations, dysarthria, and ataxia as part of the patient’s disease course. Neurologic exam revealed bilateral vertical gaze palsy, horizontal nystagmus, right-sided pronator drift, progressive akinetic mutism, and pyramidal signs such as a positive right-sided Babinski. Her initial hospital course was marked by rapidly fluctuating mental status, repetitive aphasia, and periods of agonal respirations. Non-contrast MRI of the brain was significant for a symmetrical high diffusion signal in bilateral pulvinar thalami, known as the pulvinar sign (Fig. 1). Alternative diagnoses such as Wernicke’s encephalopathy were ruled out. The history, clinical presentation, and MRI findings raised suspicion of prion disease and a lumbar puncture was performed. Routine CSF studies were within normal limits, but the sample sent to the National Prion Disease Pathology Surveillance Center (Cleveland, OH, USA) showed a positive predictive value of 72.6% for CJD based on a positive 14-3-3 level, and an elevated tau protein (1,808 pg/mL). The decision point for tau protein is 1,150 pg/mL.
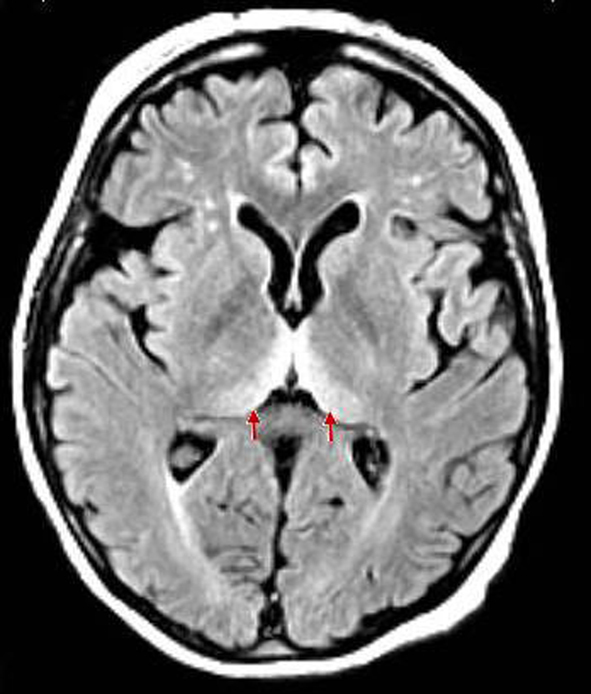 Click for large image | Figure 1. Clear findings for bilateral hyperintensities of the posterior thalamus relative to the signal intensity of the anterior putamen (pulvinar sign). |
After 18 months removed from being a local American Indian board member, the patient is non-verbal and now entirely bed bound secondary to her worsening gait which resulted in multiple falls. One of which resulted in a fractured hip. The patient’s orientation, memory, speech, and motor control are severely compromised as well. The patient is under hospice care.
| Discussion | ▴Top |
The incidence of TSE in the United State has remained stable about 1 per 1 million persons. The disease is caused by the induction of a conformational change from the non-pathologic prion protein (PrPc) to a pathologic form (PrPSc), by sporadic (s), familial (f) or exogenous (iatrogenic (I), variant (v)) means [1]. Diagnosis of prion disease is ultimately made by tissue biopsy. Definite diagnosis can be made by neuorpathological, immunocytochemical, or Western blotting techniques, to confirm the presence of protease-resistant PrP or the presence of scrapie-associated fibrils [3]. In the past, a tissue biopsy specifically required CNS tissue. However, it is now known that vCJD has the unique property of PrPsc deposition in lymphoreticular tissue, and it is estimated that tonsillar biopsy is both 100% sensitive and 100% specific for the diagnosis for vCJD [5]. Ideally, tissue biopsy should be obtained in all patients suspected of prion disease. Because of the inability of laboratory current methods to destroy PrPsc, it is unrealistic to expect all institutions to face this danger, especially with confirmed infectivity of vCJD through blood products. Making probable and possible diagnosis of either sCJD or vCJD relies on meeting evolving diagnostic criteria that include characteristic clinical features and laboratory testing [3, 4, 6].
Cases of sCJD occur primarily in individuals aged 55 - 75 years; whereas, vCJD occurs almost exclusively in persons under 55 years; the mean age at diagnosis is 29 years. In fact, patients cannot be given a diagnosis of suspected vCJD if his or her current age or age at death is greater than 55 years [4]. The median duration of illness is 4 - 5 months for sCJD but is 13 - 14 months for vCJD [7]. Many of the initial clinical features of sCJD are non-specific. They include rapidly progressive dementia, myoclonus, visual or cerebellar signs, pyramidal/extrapyramidal sign, akinetic mutism, cognitive impairment as well as psychiatric symptoms. The clinical presentation of sCJD tends to involve dementia and early neurologic signs. In contrast, vCJD often presents as psychiatric and behavioral symptoms initially and only later progress to neurological symptoms similar to those of sCJD [8]. Our patient’s age of disease onset is typical of sCJD, but the duration of illness was comparable with that for vCJD. The clinical presentation had features of both diseases.
Once a clinical suspicion for prion disease is made, investigational modalities including MRI, CSF studies, and EEG should be done to rule out alternative diagnoses. MRI is the most useful imaging modality for CJD and has now been added to the official WHO diagnostic criteria for prion disease. MRI hyperintensities in at least two cortical regions (temporal, parietal, or occipital) or both caudate nucleus and putamen are indicative of sCJD [3]. Up to 90% of sCJD patients have increased intensity in the basal ganglia and cortical ribboning [1]. These findings were absent from our case. Rather, clear findings for bilateral hyperintensities of the posterior thalamus relative to the signal intensity of the anterior putamen (pulvinar sign) (Fig. 1) were observed. This characteristic MRI finding is seen in vCJD. Several case series have estimated the pulvinar sign to be up to 100% specific for the diagnosis of vCJD [2, 9, 10]. However, few false positive pulvinar signs have been reported for sCJD [11, 12].
CSF analyses have been an element of the diagnostic criteria for sCJD for over 12 years, while known to be very insensitive in the diagnosis of vCJD. The diagnostic usefulness of CSF in prion disease relates primarily to the detection of protein markers. The most useful protein marker in prion disease has been the 14-3-3 protein, which belongs to a large family of intracellular proteins and constitutes 1% of total protein content in brain neurons. Elevation in the 14-3-3 protein is found in conditions that have rapid and extensive neuronal destruction [13]. The 14-3-3 protein is reported as a qualitative result and it is estimated to be 94% sensitive and 84% specific in the diagnosis of sCJD, whereas the 14-3-3 protein is estimated to be only 50% sensitive in the diagnosis of vCJD [14, 15]. False-positives are mainly attributed to stroke and meningoencephalitis. Tau and s100b are two other protein markers useful in the diagnosis of sCJD, and are quantitatively measured in the CSF. A 10-year study in the UK examining the usefulness of CSF in the diagnosis of sCJD found the 14-3-3 protein to be 86% sensitive and 74% specific, tau as 81% sensitive and 85% specific, and s100b as 65% sensitive and 90% specific. They reported a combination of CSF 14-3-3 protein and elevated s100B or elevated tau and s100b has a greater positive predictive value than 14-3-3 alone [16]. Although our case finding of a positive predictive value of 72.6% for sCJD leaves room for doubt, that percentage was based on a positive 14-3-3 level and an elevation of tau protein alone. The addition of s100b would have likely increased the certainty of the diagnosis.
While studies have shown that CSF analysis is more sensitive and specific than EEG in the diagnosis of sCJD [14], EEG is estimated as 66% sensitive and 74% specific for the diagnosis of sCJD. The characteristic EEG pattern in sCJD is that of background slowing with repetitive (> 5) bi- or triphasic periodic complexes, occurring at 0.5 - 2 s with < 0.5 s variability between complexes and distributed widely over the cortex [14]. The prevalence of the characteristic periodic complexes increases with the age of the patient and decreases with the disease duration. The likelihood of the characteristic EEG findings also varies with the genotype of prion disease. It is these and other unknown factors that could have contributed to a non-specific EEG as evident in our case. EEG studies, however, are notoriously negative in vCJD [14, 17].
Our case shares features of both sCJD and vCJD (Table 1) and fulfills WHO criteria for probable diagnosis of sCJD and suspected vCJD due to the pulvinar sign on MRI. While the non-specific EEG and MRI findings support suspected vCJD, our patient’s age of onset and positive 14-3-3 and tau CSF protein markers do not fit what we know about vCJD and thus, favor a diagnosis of probable sCJD more strongly.
 Click to view | Table 1. WHO’s Diagnostic Criteria for Creutzfeldt-Jakob Disease, 2010 |
Conclusion
The authors present a notable case of an individual who by using the WHO criteria is given a probable diagnosis of sCJD while exhibiting MRI findings that are typically seen in vCJD. Our case exemplifies commonality between sCJD and vCJD and challenges the utility of radiological findings differentiating the diseases. Our case emphasizes that the pulvinar sign may not be as specific for a diagnosing vCJD as we once thought. We propose that this case join one of few reported cases of sCJD masking as vCJD.
Disclosure
Poster presented at OU-tulsa Clinical Vignette Symposium on April 26, 2012. Tulsa, Oklahoma.
| References | ▴Top |
- Prusiner B, et al. Prion Disease. Harrison's principles of internal medicine (18th ed). New York, NY: McGraw Hill. 2012;3441-3447.
- Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, Zeidler M, Knight R, et al. Diagnosing variant Creutzfeldt-Jakob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. AJNR Am J Neuroradiol. 2003;24(8):1560-1569.
pubmed - World Health Organisation. Diagnosis and Therapy of Human Transmissible Spongiform Encephalopathies: Report of a WHO Consultation. Geneva, Switzerland, 1998. World Health Organisation. Diagnosis and Therapy of Human Transmissible Spongiform Encephalopathies: Report of a WHO Consultation. Geneva, Switzerland, 1998.
- World Health Organization. The Revision of the Surveillance Case Definition for Variant Creutzfeldt-Jakob Disease (vCJD):[WHO/CDS/CSR/EPH/2001.5]. Geneva, Switzerland, 2001. World Health Organization. The Revision of the Surveillance Case Definition for Variant Creutzfeldt-Jakob Disease (vCJD):[WHO/CDS/CSR/EPH/2001.5]. Geneva, Switzerland, 2001.
- Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, Thomas DJ, Frosh A, et al. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet. 1999;353(9148):183-189.
doi - Ironside JW, McCardle L, Horsburgh A, Lim Z, Head MW. Pathological diagnosis of variant Creutzfeldt-Jakob disease. APMIS. 2002;110(1):79-87.
doi pubmed - Belay ED, Schonberger LB. Variant Creutzfeldt-Jakob disease and bovine spongiform encephalopathy. Clin Lab Med. 2002;22(4):849-862, v-vi.
doi - Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213-239.
doi pubmed - Lodi R, Parchi P, Tonon C, Manners D, Capellari S, Strammiello R, Rinaldi R, et al. Magnetic resonance diagnostic markers in clinically sporadic prion disease: a combined brain magnetic resonance imaging and spectroscopy study. Brain. 2009;132(Pt 10):2669-2679.
doi pubmed - Zeidler M, Sellar RJ, Collie DA, Knight R, Stewart G, Macleod MA, Ironside JW, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease. Lancet. 2000;355(9213):1412-1418.
doi - Petzold GC, Westner I, Bohner G, Einhaupl KM, Kretzschmar HA, Valdueza JM. False-positive pulvinar sign on MRI in sporadic Creutzfeldt-Jakob disease. Neurology. 2004;62(7):1235-1236.
doi pubmed - Haik S, Brandel JP, Oppenheim C, Sazdovitch V, Dormont D, Hauw JJ, Marsault C. Sporadic CJD clinically mimicking variant CJD with bilateral increased signal in the pulvinar. Neurology. 2002;58(1):148-149.
doi pubmed - Torres M, Cartier L, Matamala JM, Hernandez N, Woehlbier U, Hetz C. Altered Prion protein expression pattern in CSF as a biomarker for Creutzfeldt-Jakob disease. PLoS One. 2012;7(4):e36159.
doi pubmed - Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS, Bernheimer H, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000;55(6):811-815.
doi pubmed - Green AJ, Thompson EJ, Stewart GE, Zeidler M, McKenzie JM, MacLeod MA, Ironside JW, et al. Use of 14-3-3 and other brain-specific proteins in CSF in the diagnosis of variant Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2001;70(6):744-748.
doi pubmed - Chohan G, et al. The role of protein 14-3-3 and other proteins in CSF and diagnosis of sporadic Creutzfeldt-Jakob Disease in the UK: a 10-year review. J Neurol Neurosurg Psychiatry. 2010;81(11):1243-1248.
doi pubmed - Holman RC, Belay ED, Christensen KY, Maddox RA, Minino AM, Folkema AM, Haberling DL, et al. Human prion diseases in the United States. PLoS One. 2010;5(1):e8521.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.