

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 4, Number 2, February 2013, pages 92-98
Reversible Myocardial Calcification Following Severe Leptospirosis Complicated With Rhabdomyolysis-Induced Acute Kidney Injury and Magnesium-Wasting Nephropathy
Raed Zakouta, d, Maria do Carmo Perloirob, Paulo Freitasc
aDepartement of Medicine I, Hospital Prof. Doutor Fernando Fonseca, E.P.E., Portugal
bDepartement of Medicine II, Hospital Prof. Doutor Fernando Fonseca, E.P.E., Portugal
cIntensive Care Unit, Hospital Prof. Doutor Fernando Fonseca, E.P.E., Portugal
dCorrespondent author: Raed Zakout, Department of Medicine I, Hospital Prof. Doutor Fernando Fonseca, E.P.E., 2720-276 Amadora, Portugal
Manuscript accepted for publication October 24, 2012
Short title: Reversible Myocardial Calcification
doi: https://doi.org/10.4021/jmc943w
| Abstract | ▴Top |
We present a patient with leptospirosis infection who presented septic shock with multiple-organ dysfunction syndrome, severe rhabdomyolysis and acute myocarditis. He developed biphasic blood calcium pattern with hypocalcemia in the oliguric phase followed by hypercalcemia during the recovery diuretic phase in the context of rhabdomyolysis and oliguric acute kidney injury. Meanwhile, he developed an extensive calcification of the myocardium. Severe renal magnesium wasting was observed during the convalescence phase. Follow-up showed progressive resorption and later almost total disappearance of the calcific deposits in the heart by the 18th month after discharge. Renal magnesium wasting decreased gradually, but yet persisted beyond the 18th and was normalized only by the 36th month after discharge. We discuss the pathophysiologic mechanisms involved in the myocardial calcification and renal magnesium wasting and suggest a possibility of a contributing role of magnesium renal wasting in mobilization of calcium deposits out of myocardium.
Keywords: Leptospirosis; Magnesium wasting; Acute kidney injury; Myocardial calcification; Rhabdomyolysis; Tubular dysfunction
| Introduction | ▴Top |
Leptospirosis is an important zoonotic emerging disease caused by Leptospira interrogans that occurs worldwide. Rats are the most common vector for human infection. Infected animals shed L. interrogans into their urine. Young adult men often become infected, for example, after recreational exposure to infected animal urine-contaminated water or soil. Leptospirosis varies from subclinical illness to severe potentially fatal disease associated with septic shock and multiple organ failure. Severe rhabdomyolysis and potentially fatal acute myocarditis are other important complications of leptospirosis. Renal involvement is a cardinal feature of leptospirosis and it ranges from polyuria and solute wasting to severe acute kidney injury requiring dialysis. This case report describes a patient with severe leptospirosis that was complicated with reversible extensive myocardial calcification and hypomagnesemic renal magnesium wasting that persisted for several months.
| Case Report | ▴Top |
A previously healthy 40-year-old man was admitted to the hospital with a 3-day history of fever, malaise, anorexia, myalgia, arthralgia, vomiting and diarrhea. He referred recreational activity in his home garden following rainy days 10 days before the admission. On physical examination he was febrile and dehydrated. The rest of physical examination was unremarkable. Initial laboratory evaluation revealed hemoglobin 13.2 g/dL, white blood cell count (WBC) 12.9 × 109/L with neutrophils 11.4 × 109/L, platelets 35 × 109/L, PCR 39.2 mg/dL, AST 41 U/L, ALT 65 U/L, total bilirubin 0.6 mg/dL, LDH 157 U/L, CK 332 U/L, sodium 134 mmol/L, potassium 3.9 mmol/L, blood urea nitrogen (BUN) 38 mg/dL and creatinine 4.4 mg/dL. Urinalysis was normal. Chest X-ray showed bilateral interstitial infiltrates. He initiated intravenous fluid therapy, amoxyciline-clavulanate and clarythromycine after the collection of three blood cultures and one urine culture that later yielded negative results. The patient rapidly developed septic shock with multiple-organ dysfunction syndrome including circulatory failure, acute lung injury, hepatic failure, and the acute kidney injury (AKI) had progressed to oliguria. He was intubated and transferred to the intensive care unit (ICU) on the 2nd hospitalization day. Laboratory evaluation at that point was as follows: WBC 25.6 × 109/L, neutrophils 21.9 × 109/L, PCR 39.4 mg/dL, prothrombin time 19.7 s (INR 1.8), albumin 2.5 mg/dL, AST 14,020 U/L, total bilirubin 18 mg/dL, LDH 7,680 U/L, CK 100,000 U/L, CK-MB 2,140 U/L, troponine I 123 ng/mL, sodium 130 mmol/L, potassium 4.8 mmol/L, BUN 165 mg/dL, creatinine 5.4 mg/dL, magnesium 2.7 mg/dL, albumin-corrected calcium 7.8 mg/dL, phosphorus 6.7 mg/dL. Three sets of two blood cultures were again negative. Serologic study for Mycoplasma pneumoniae, Chlamydia pneumoniae, Chlamydia psittaci, Legionella pneumophila, Borrelia burgdorferi, Rickettsia conorii, Rickettsia rickettsii, Rickettsia typhi, cytomegalovirus, Ebstein-Barr virus, hepatitis B virus, hepatitis C virus and human immunodeficiency virus was negative. Indirect hemagglutination test for leptospirosis (Focus Diagnostics, CA, USA) was also negative. Repeated serology for leptospirosis 2 weeks later revealed seroconversion with titer 1/800. Electrocardiogram revealed sinus rhythm, right bundle-branch block (RBBB) pattern and low voltage QRS complexes. In the ICU the patient started empiric antibiotic therapy with meropenem and continued clarythromicine. Perfusion of dopamine and noradrenaline was also started for four and seven days, respectively. He was also on continuous veno-venous hemodiafiltration for 11 days, and thereafter on intermittent hemodialysis (IHD) for another 16 days. He was extubated on the 10th day in the ICU. After the serologic confirmation of leptospirosis, the patient was de-escalated to ceftriaxone and doxycycline. During renal replacement therapy and particularly in the first few days after the switch to IHD, he had persistent mild albumin-corrected hypocalcemia with the need for perfusion of calcium gluconate. When IHD was stopped on the 27th day in ICU, serum creatinine was 3.4 mg/dL and albumin-corrected calcium was 10.5 mg/dL. Two days after IHD had been stopped; the patient was transferred to medicine ward, where he developed progressive polyuria, hypercalcemia and hypomagnesemia, with the worst parameters were 6 L/day, 15.4 mg/dL and 1.4 mg/dL respectively on around the 10th day after IHD was stopped, with serum creatinine had been decreased to 1.2 mg/dL (Fig. 1). Mild hypokalemia and metabolic alkalosis were also observed during this diuretic phase. The study of parathyroid hormone (PTH)-vitamin D axis at that time showed: PTH 3.57 pg/mL (normal range 12 - 65), 25-hydroxyvitamin D 29 ng/mL (n 12 - 80), and 1, 25-dihydroxivitamin D (1, 25-(OH)2D) < 2.8 pg/mL (n 18 - 78). At that point, linear calcification on the left border of the cardiac silhouette was noticed on chest x-ray (Fig. 2A). CT scan showed calcification of left ventricle, parts of right ventricle, and interventricular septum (Fig. 2B), and diffuse calcification of pelvic and psoas muscles. Kidneys were normal and with no evidence of nephrolithiasis. Initial transthoracic echocardiogram showed mild left ventricular (LV) dilation and impaired global LV systolic function (ejection fraction = 45%), with hypokinesia of inferior and posterior walls. Trans-mitral flow velocity and mitral annular velocity made constrictive pericarditis unlikely. Cardiac magnetic resonance showed typical pattern of delayed enhancement distribution suggestive of myocarditis and virtually excluded myocardial infarction. He was medicated with pamidronate and fluids. He also had been on intravenous magnesium sulphate (6 g/day) and potassium chloride. Furosemide was not administered because of the high urinary output. Calcium returned toward normal values some days after pamidronate administration. Urinary output returned to normal 3 weeks after IHD was stopped. After urinary output returned to normal, serum potassium and acid-base balance normalized, but hypomagnesemia persisted. Laboratory evaluation 4 weeks after IHD was stopped, while yet on high doses of intravenous magnesium and out of potassium chloride, showed elevated urinary magnesium excretion in the presence of hypomagnesemia, suggesting renal wasting of magnesium, and also showed severe hypercalciuria despite serum calcium had returned to normal (Table 1). He was discharged treated with oral magnesium pidolate (4.5 g/day) and angiotensin-receptor inhibitor. Serial measurements showed gradual decrease of magnesiuria and so magnesium supplement was tapered. On the 18th month after discharge and while yet on oral magnesium pidolate 1.5 g/day; magnesium was 1.7 mg/dL and FEMg 6.8% (Table 1). On the 36th month after discharge, with magnesium supplement had been stopped several months earlier; magnesium was 1.9 mg/dL and FEMg 2.0% (Table 1). Serial echocardiograms after discharge showed progressive recovery of global and regional cardiac function, maintaining mild LV dilatation (EDd = 60 mm). Echocardiogram performed at 36th month after discharge, showed normal values of regional strain and strain rate, namely in the segments of the inferior and posterior walls. Serial CT scans showed progressive resorption and on the 18th month after discharge, almost total disappearance of calcific deposits in the heart (Fig. 2C). CT scan on the 36th month after discharge showed only millimetric calcification in the left ventricle (Fig. 2D). Electrocardiogram at this time showed RBBB, but was otherwise normal.
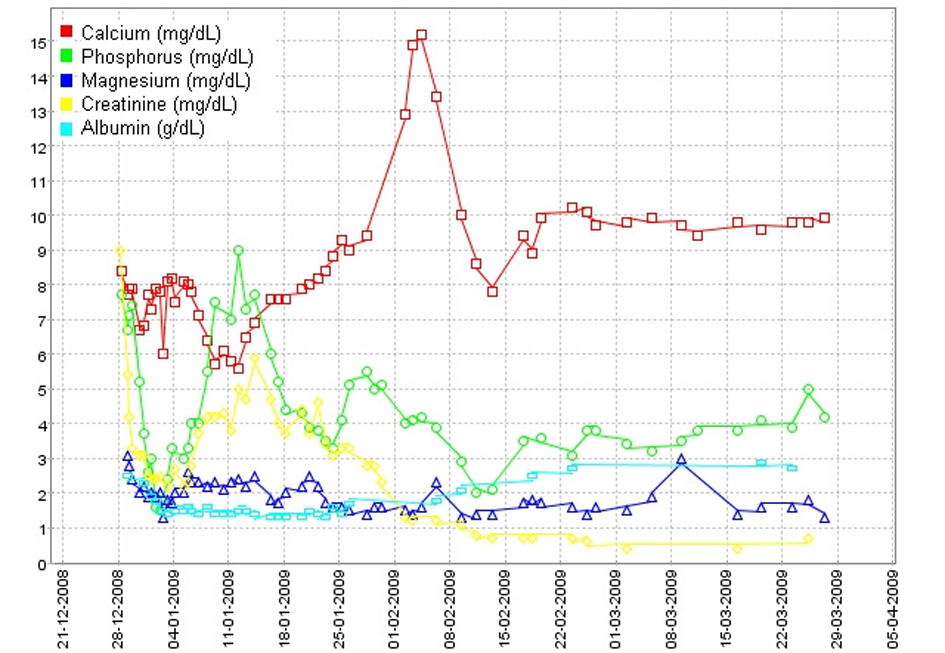 Click for large image | Figure 1. Evolution of serum calcium, phosphorus, magnesium, creatinine and albumin levels during the hospital stay and first weeks after discharge. |
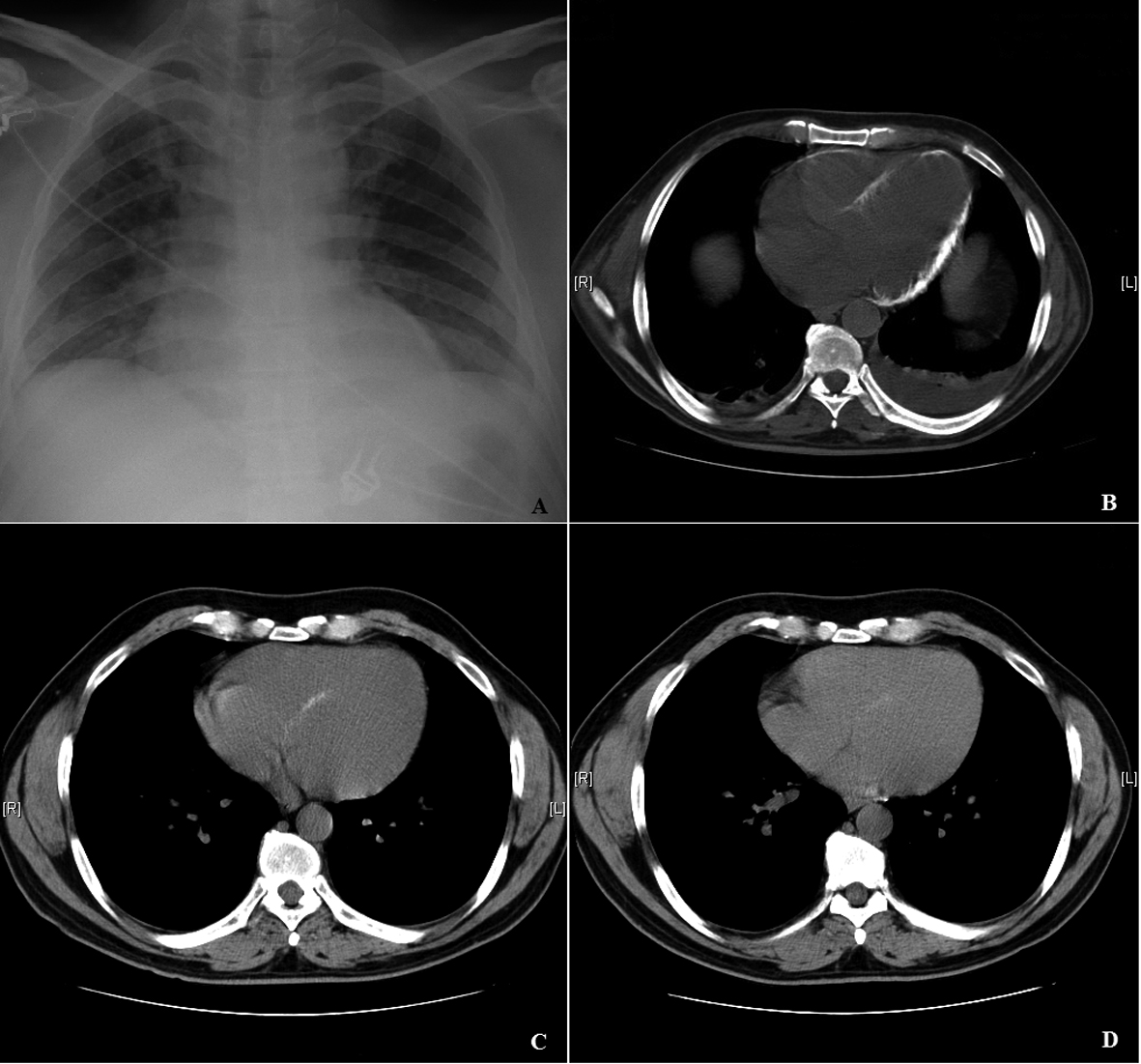 Click for large image | Figure 2. A: Chest x-ray: Linear calcification on the left border of the cardiac silhouette. B: CT scan: Calcification of left ventricle, parts of right ventricle, and interventricular septum. C: CT scan by the 18th month after discharge: Progressive resorption of calcific deposits in the heart. D: CT scan by the 36th month after discharge: Only millimetric calcification in the left ventricle. |
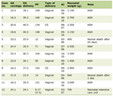 Click to view | Table 1. Laboratory Evaluation Results |
| Discussion | ▴Top |
Leptospirosis is an emerging infectious disease characterized by a broad spectrum of clinical manifestations ranging from unapparent infection to fulminant disease associated with multiple organ failure. In our patient, the peculiar clinical context of severe sepsis, myocarditis, rhabdomyolysis-associated acute kidney injury (AKI) and possibly vasopressor therapy promoted calcification of the heart. The patient also developed hypomagnesemia due to renal magnesium wasting that was yet observed 18 months after the recovery of renal function, in dissimilar from other reported cases, where magnesium wasting in leptospirosis was observed in the acute phase and completely reversed during the convalescence phase.
Calcification refers to the pathologic deposition of calcium salts in soft tissues. Pathologic calcification can be classified as either metastatic, in which calcium deposits in normal tissues, or dystrophic, in which calcification occurs in previously injured tissues. Metastatic calcification is seen in patients on chronic hemodialysis for chronic renal failure, whereas causes of dystrophic calcification include, for example, myocardial calcification after myocardial infarction or myocarditis. In rhabdomyolysis-induced AKI, temporary ectopic calcification in affected skeletal muscles is common and it can decrease or even disappear within few months [1-3]. Biphasic calcemic pattern observed here, with hypocalcemia in the oliguric phase followed by hypercalcemia during the recovery diuretic phase is characteristic for rhabdomyolysis-associated AKI. This sequence reflects widespread deposition of calcium in damaged muscle and subsequent mobilization of the calcium out from the recovering muscle as phosphorous and creatinine values return to normal [1]. The combination of increased release of phosphate from injured muscle and impaired ability to excrete phosphorous because of renal failure causes hyperphosphatemia, the latter causing hypocalcemia due to calcium phosphate precipitation. Decreased production of 1, 25(OH)2D by the impaired kidneys and resistance of injured muscle to parathyroid hormone are believed to be other possible mechanisms of hypocalcemia [4]. The fall of phosphate levels after restoration of renal function causes release of deposited calcium to the extracellular fluid [4]. Conflicting results implicate the parathyroid hormone in these calcemic alterations [5]. However, low PTH and 1, 25(OH)2D in our patient during the hypercalcemic phase could argument that there was no disruption in the PTH-vitamin D axis regulating calcium levels, and suggest that low PTH was secondary to altered calcium levels [4].
Myocardial calcification has been rarely reported in septic shock, despite the high incidence of the later [6-11]. Myocardial calcification has been also described in acute myocarditis [12, 13]. Those include a case of massive myocardial calcification after acute myocarditis attributed to an unidentified infection, complicated with septic shock and rhabdomyolysis-induced AKI, where the calcification had been gradually diminished in the next 18 months of follow-up [13]. Although elevation of cardiac troponin is well documented in sepsis and correlates with the severity of this disease, the extremely high levels of cardiac enzymes on admission and cardiac MRI suggest myocarditis. It seems that the myocardial cells injury and associated oliguric AKI promoted the precipitation of calcium in the myocardium. Although precipitation of calcium in inflamed myocardium during the hypercalcemic phase is a tentative explanation, there is evidence that calcification might had occurred during the hypocalcemia [13, 14]. Moreover, it was also observed in patients without a hypercalcemic phase [13]. Disappearance of myocardial calcifications in subsequent image studies demonstrates that they are temporary and parallel to the activity of myocarditis [15]. Although there was improvement of cardiac function in subsequent echocardiograms, dilated cardiomyopathy as a consequence of severe leptospiral myocarditis had occurred.
Calcium administration during the hyperphosphatemic period tends to increase extraosseous calcium deposition and aggravate tissue damage. To minimize the risk of calcium deposition and delayed hypercalcemia, correction of hypocalcemia in rhabdomyolysis-associated AKI should be avoided, unless symptomatic or associated with severe hyperkalemia. In fact, some authors consider ICU associated hypocalcemia as a protective and beneficial response to critical illness, as it may prevent intracellular hypercalcemia and associated tissue damage [16].
Another interesting aspect in this case is the prolonged renal magnesium wasting. Tubulointersticial nephritis is the cardinal manifestation of AKI in liptospirosis; and is usually characterized by polyuria, sodium and potassium renal wasting, and hypokalemia [17]. However, oliguria is described in up to 11-24% of patients and it is higher among severe forms [18, 19]. Dehydration, rhabdomyolysis and jaundice are other involved factors that cause progression to oliguric AKI [19, 20]. The pathophysiologic mechanisms of polyuria and electrolytes wasting were shown by clinical and experimental studies to be caused by direct leptospiral inhibitory action on tubular cells sodium transporters, resulting in more distal sodium delivery and subsequent renal potassium loss due to its increased secretion in the collecting duct [17, 21-29]. Sodium-potassium-ATPase (Na-K-ATPase) dysfunction has been described in the whole nephron or just in the proximal tubule [17, 21, 24, 25, 29]. This would be expected to reduce the drive through sodium transporters in the apical membrane; the proximal tubule type-3 sodium-hydrogen exchanger (NHE3) and the sodium-potassium-chloride co-transporter (NKCC2) in the thick ascending limb of Henle’s loop (TAL). Defective function and decreased expression of NHE3 was shown in human and experimental studies [21, 22, 29, 30]. Impairment of NHE3 function would explain the occurrence of proximal renal tubular acidosis in leptospirosis [30]. Detailed in vivo tubular clearance tests were performed in serovar Shermani infected patients and identified dysfunction of NKCC2 [27]. In vitro studies demonstrated downregulation of NKCC2 activity in the TAL segment by serovars Shermani and Copenhageni [22, 28]. However, other serovars were shown not to affect or even to up-regulate the function of NKCC2, apparently as a compensatory response to the greater sodium and water delivery by the proximal tubule due to inhibition of NHE3 [21]. Diminished expression of aquaporin 1 in the proximal convoluted tubule was documented and this might contribute to deficient water reabsorption and, therefore, to polyuria [29]. The controversy about the tubular dysfunction might reflect the injury of miscellaneous nephron segments, depending on leptospiral load, serogroup or examination in different phases of illness [27, 31]. Other causes of tubular damage indirectly related to leptospira, such as sepsis, tubular ischemia/necrosis, hyperbilirubinemia and myoglobinuria might contribute to the diversity of tubular defects. Proximal tubular defects other than impaired sodium reabsorption have been reported, as demonstrated by reports of reversible proximal renal tubular acidosis, glycosuria, hyperphosphaturia and hyperuricosuria [30, 32, 33]. Incomplete proximal renal tubular acidosis was observed in 10% of leptospirosis patients with AKI [26]. Cellular transport of most solutes in the proximal tubule is coupled to sodium concentration gradient established by the activity of basolateral Na-K-ATPase. Therefore, inhibition of Na-K-ATPase would cause renal solutes loss by the apical Na-dependent transporters such as Na-glucose and Na-phosphate contransporters. Regardless of the tubule segment involved, leptospiral inhibition of tubular cells sodium transporters is expected to lead to hypermagnesiuria. For instance, inhibition of NKCC2 causes reduction of transepithelial voltage and as a result reduces the passive paracelullar magnesium reabsorption, leading to renal magnesium loss [28]. In fact, hypomagnesemic renal magnesium wasting in leptospirosis has been described in recent literature [34-36]. A clinical case report of severe hypomagnesemia secondary to renal wasting, requiring large doses of magnesium replacement during the acute phase of leptospirosis associated with non-oliguric AKI was reported [35]. A series of 20 patients with leptospirosis reported elevated FEMg in 15 of them that was more severe and caused hypomagnesemia in patients who also had hypovolemic shock and oliguric AKI on admission [34]. Evidence of proximal tubule damage was observed in all these patients, including hyperphosphaturia in 50% of them, but these alterations regressed to normal within 2 weeks. Urinary calcium excretion and the activity of the Na-Cl co-transporter in the distal convoluted tubule, a transporter which involvement could lead to renal magnesium wasting, were normal in all patients [34]. Dysfunction of TAL was suggested to be the likely cause of hypermagnesiuria [34]. Due to absence of hypokalemic metabolic alkalosis, Khositseth et al suggested that dysfunction of NKCC2 could not explain renal magnesium wasting in his patients and proposed direct leptospiral damage to renal tight junction protein paracellin-1 (claudin 16) as a possible mechanism [34]. In TAL, claudins 16 and 19 form the pore permitting paracellular magnesium reabsorption, a process which depends on the transepithelial voltage which is, nevertheless, maintained by the activity of NKCC2. In these reports, hypermagnesiuria was transitory during the acute phase and regressed or significantly improved in few days.
Recovery of glomerular and tubular renal function is early and almost complete even in severe forms of leptospirosis [18, 19]. However, a clinical case of a patient with dialysis-requiring-AKI who did not recover his renal function and remained dialysis-dependent was reported [37]. In a study by Covic et al, tubular dysfunction persisted after a 3-month follow up in 30% of patients with severe leptospirosis who completely recovered their glomerular function [26]. In another series of 35 patients by Daher et al, proximal sodium reabsorption and FENa recovered but elevated FEK and impaired urinary concentration ability yet persisted on the 3rd month after discharge; however, by the 6th month FEK returned to normal but urinary concentration ability remained altered [19]. In the series by Khositseth et al, hypermagnesiuria normalized or significantly improved in few days, with FEMg decreasing from values as high as 43.8% on admission to 4.5% after 2 weeks [34]. Initially, renal magnesium wasting in our patient could be attributed to diuretic phase of renal function recovery and to hypercalciuria. However, persistence of renal magnesium wasting after the patient had both urinary output and renal calcium excretion normalized, suggested that it was secondary to leptospirosis-associated tubular dysfunction. Our patient had not been treated with diuretics. The absence of metabolic alkalosis or hypokalemia, and the normal FEk and FENa in the follow-up evaluation realized on the 18th and 36th month made unlikely Bartter’s and Gitelman’s syndromes. Moreover, the serial laboratorial evaluation during the extended follow-up period showed gradual decrease in the magnitude and later normalization of magnesium renal excretion, proving definitely the relationship of hypermagnesiuria to leptospirosis infection, and excluding hereditary hypomagnesemic syndromes. Unlike the previous described reports, renal magnesium wasting and hypomagnesemia in our patient persisted for several months and long-term oral magnesium was prescribed to maintain normal magnesium. We hypothesize that renal magnesium wasting might contributed to the regression of the myocardial calcification. It is possible that renal magnesium wasting had contributed to increased calcium urinary excretion and mobilization of deposited calcium as high urinary magnesium may compete for reabsorption with calcium at the TRPV5 channel in the distal convoluted and connecting tubule [38].
Acknowledgments
We would like to acknowledge Dr. Antonio Freitas, Department of Cardiology, Hospital Prof. Doutor Fernando Fonseca, E.P.E., for his contribution in the revision and discussion of echocardiograms and cardiac magnetic resonance and Dr. Pedro Cabral, Department of Radiology, Hospital Prof. Doutor Fernando Fonseca, E.P.E., for his contribution in the revision of CT scans.
Conflict of Interest
No conflict of interest is declared.
| References | ▴Top |
- Akmal M, Goldstein DA, Telfer N, Wilkinson E, Massry SG. Resolution of muscle calcification in rhabdomyolysis and acute renal failure. Ann Intern Med. 1978;89(6):928-930.
pubmed - Towers MJ, Downey DB, Poon PY. Psoas muscle calcification and acute renal failure associated with nontraumatic rhabdomyolysis: CT features. J Comput Assist Tomogr. 1990;14(6):1027-1029.
doi pubmed - Sasaki H, Ishii K, Kono AK, Miyamoto N, Fukuda T, Kanata J, Kita Y. Early disappearance of calcification in posterior paraspinal muscles in a patient with rhabdomyolysis associated with neuroleptic malignant syndrome. Radiat Med. 2006;24(6):463-466.
doi pubmed - Shrestha SM, Berry JL, Davies M, Ballardie FW. Biphasic hypercalcemia in severe rhabdomyolysis: serial analysis of PTH and vitamin D metabolites. A case report and literature review. Am J Kidney Dis. 2004;43(3):e31-35.
doi pubmed - Llach F, Felsenfeld AJ, Haussler MR. The pathophysiology of altered calcium metabolism in rhabdomyolysis-induced acute renal failure. Interactions of parathyroid hormone, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol. N Engl J Med. 1981;305(3):117-123.
doi pubmed - Lapatto-Reiniluoto O, Vaalamo M, Takkunen O, Manttari M. Left ventricular calcification following resuscitation. J Intern Med. 2000;248(1):85-87.
doi pubmed - Schellhammer F, Ansen S, Arnold G, Brochhagen HG, Lackner K. Myocardial calcification following septic shock. Cardiology. 2002;98(1-2):102-103.
doi pubmed - Rossi MA, Santos CS. Sepsis-related microvascular myocardial damage with giant cell inflammation and calcification. Virchows Arch. 2003;443(1):87-92.
doi pubmed - Sinicina I, Matevossian E, Fischer F, Mall G, Graw M. The petrified heart in sepsis. Virchows Arch. 2005;447(5):875-878.
doi pubmed - Simonson S, Miller WT, Jr., Perl A, Torigian DA. Diffuse left ventricular myocardial calcification in the setting of sepsis on CT imaging. J Thorac Imaging. 2007;22(4):343-345.
doi pubmed - van Kruijsdijk RC, van der Heijden JJ, Uijlings R, Otterspoor LC. Sepsis-related myocardial calcification. Circ Heart Fail. 2011;4(5):e16-18.
doi pubmed - Itoh E, Saitoh H, Miida T, Oda H, Toeda T, Higuma N, Okazaki E. An autopsied case of acute myocarditis with myocardial calcification. Jpn Circ J. 1997;61(9):798-802.
doi pubmed - Wada A, Nakata T, Tsuchihashi K, Aoyama S, Nanba M, Murakami H, Shimamoto K, et al. Massive myocardial calcification of right and left ventricles following acute myocarditis complicated with rhabdomyolysis-induced acute renal failure. Jpn Circ J. 1993;57(6):567-572.
doi pubmed - Akmal M, Bishop JE, Telfer N, Norman AW, Massry SG. Hypocalcemia and hypercalcemia in patients with rhabdomyolysis with and without acute renal failure. J Clin Endocrinol Metab. 1986;63(1):137-142.
doi pubmed - Lane JT, Boudreau RJ, Kinlaw WB. Disappearance of muscular calcium deposits during resolution of prolonged rhabdomyolysis-induced hypercalcemia. Am J Med. 1990;89(4):523-525.
doi - Zaloga GP. Ionized hypocalcemia during sepsis. Crit Care Med. 2000;28(1):266-268.
doi pubmed - Seguro AC, Lomar AV, Rocha AS. Acute renal failure of leptospirosis: nonoliguric and hypokalemic forms. Nephron. 1990;55(2):146-151.
doi pubmed - Daher E, Zanetta DM, Cavalcante MB, Abdulkader RC. Risk factors for death and changing patterns in leptospirosis acute renal failure. Am J Trop Med Hyg. 1999;61(4):630-634.
pubmed - Daher Ede F, Zanetta DM, Abdulkader RC. Pattern of renal function recovery after leptospirosis acute renal failure. Nephron Clin Pract. 2004;98(1):c8-14.
doi pubmed - Abdulkader RC. Acute renal failure in leptospirosis. Ren Fail. 1997;19(2):191-198.
doi pubmed - Andrade L, Rodrigues AC, Jr., Sanches TR, Souza RB, Seguro AC. Leptospirosis leads to dysregulation of sodium transporters in the kidney and lung. Am J Physiol Renal Physiol. 2007;292(2):F586-592.
doi pubmed - Spichler A, Ko AI, Silva EF, De Brito T, Silva AM, Athanazio D, Silva C, et al. Reversal of renal tubule transporter downregulation during severe leptospirosis with antimicrobial therapy. Am J Trop Med Hyg. 2007;77(6):1111-1119.
pubmed - Yang CW, Wu MS, Pan MJ, Hong JJ, Yu CC, Vandewalle A, Huang CC. Leptospira outer membrane protein activates NF-kappaB and downstream genes expressed in medullary thick ascending limb cells. J Am Soc Nephrol. 2000;11(11):2017-2026.
pubmed - Magaldi AJ, Yasuda PN, Kudo LH, Seguro AC, Rocha AS. Renal involvement in leptospirosis: a pathophysiologic study. Nephron. 1992;62(3):332-339.
doi pubmed - Younes-Ibrahim M, Buffin-Meyer B, Cheval L, Burth P, Castro-Faria MV, Barlet-Bas C, Marsy S, et al. Na,K-ATPase: a molecular target for Leptospira interrogans endotoxin. Braz J Med Biol Res. 1997;30(2):213-223.
doi pubmed - Covic A, Goldsmith DJ, Gusbeth-Tatomir P, Seica A, Covic M. A retrospective 5-year study in Moldova of acute renal failure due to leptospirosis: 58 cases and a review of the literature. Nephrol Dial Transplant. 2003;18(6):1128-1134.
doi pubmed - Lin CL, Wu MS, Yang CW, Huang CC. Leptospirosis associated with hypokalaemia and thick ascending limb dysfunction. Nephrol Dial Transplant. 1999;14(1):193-195.
doi pubmed - Wu MS, Yang CW, Pan MJ, Chang CT, Chen YC. Reduced renal Na+-K+-Cl- co-transporter activity and inhibited NKCC2 mRNA expression by Leptospira shermani: from bed-side to bench. Nephrol Dial Transplant. 2004;19(10):2472-2479.
doi pubmed - Araujo ER, Seguro AC, Spichler A, Magaldi AJ, Volpini RA, De Brito T. Acute kidney injury in human leptospirosis: an immunohistochemical study with pathophysiological correlation. Virchows Arch. 2010;456(4):367-375.
doi pubmed - Yang CW, Pan MJ, Wu MS, Chen YM, Tsen YT, Lin CL, Wu CH, et al. Leptospirosis: an ignored cause of acute renal failure in Taiwan. Am J Kidney Dis. 1997;30(6):840-845.
doi - Yang CW, Wu MS, Pan MJ. Leptospirosis renal disease. Nephrol Dial Transplant. 2001;16(Suppl 5):73-77.
doi pubmed - Liamis G, Rizos E, Elisaf MS. Reversible proximal tubular dysfunction in a patient with acute febrile illness and normal renal function: an evidence towards leptospirosis. Clin Nephrol. 2000;53(4):316.
pubmed - Liberopoulos E, Bairaktari E, Elisaf M. Reversible proximal tubular dysfunction in a patient with acute febrile illness, marked hyperbilirubinemia and normal renal function: evidence of leptospirosis. Nephron. 2002;91(3):532-533.
doi pubmed - Khositseth S, Sudjaritjan N, Tananchai P, Ong-ajyuth S, Sitprija V, Thongboonkerd V. Renal magnesium wasting and tubular dysfunction in leptospirosis. Nephrol Dial Transplant. 2008;23(3):952-958.
doi pubmed - Spichler A, Athanazio DA, Furtado J, Seguro A, Vinetz JM. Case report: severe, symptomatic hypomagnesemia in acute leptospirosis. Am J Trop Med Hyg. 2008;79(6):915-917.
pubmed - Craig SB, Graham GC, Burns MA, Dohnt MF, Jansen CC, Smythe LD, McKay DB. Hypomagnesaemia in the first 10 days of severe leptospirosis. Ann Trop Med Parasitol. 2009;103(8):705-709.
doi pubmed - Atasoyu EM, Turhan V, Unver S, Evrenkaya TR, Yildirim S. A case of leptospirosis presenting with end-stage renal failure. Nephrol Dial Transplant. 2005;20(10):2290-2292.
doi pubmed - Bonny O, Rubin A, Huang CL, Frawley WH, Pak CY, Moe OW. Mechanism of urinary calcium regulation by urinary magnesium and pH. J Am Soc Nephrol. 2008;19(8):1530-1537.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.