

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 3, Number 2, April 2012, pages 155-159
Hypersensitivity Pneumonitis: A Clinical Case
Patricia Alvesa, c, Marco Simoesa, Helder Espertoa, Elisa Meiraa, Erica Ferreiraa, Manuel Batistaa, Fernando Santosa, Jose Nascimento Costaa, b
aInternal Medicine Department, Hospitais da Universidade de Coimbra, Portugal
bFaculdade de Medicina da Universidade de Coimbra, Portugal
cCorresponding author: Patricia Alves, Rua Quinta da Varzea, Lote 6-n°2, 1°B, 3040-380 Coimbra, Portugal
Manuscript accepted for publication January 27, 2012
Short title: Hypersensitivity Pneumonitis
doi: https://doi.org/10.4021/jmc537w
| Abstract | ▴Top |
The hypersensitivity pneumonitis (HP) or extrinsic allergic alveolitis (EAA) is the paradigm of lung response to inhaled organic and inorganic. It is a relatively rare disease, constituting 2% of cases of interstitial lung diseases.The authors describe a case of a 50 years old man, with occupational exposure to metals and paints, who presented with weakness, easy fatigability on small and medium exertion, nonproductive cough, dyspnea and not quantified weight loss. Physical examination revealed bilateral crackles. The study pointed to the initial diagnosis of sarcoidosis with severe restrictive syndrome, but further study revealed that it was of hypersensitivity pneumonitis by lipid inhalation (paint-related products and lead). The patient underwent treatment with prednisone with poor response, showing improvement only after introduction of azathioprine.The authors present this case to emphasize the importance of history of exposure to any toxic substances, a lack of specificity of clinical manifestations, the need for invasive diagnostic methods and, sometimes, poor response to therapy.
Keywords: Hypersensitivity pneumonitis; Lipid inhalation; Lung biopsy
| Introduction | ▴Top |
The hypersensitivity pneumonitis (HP) or extrinsic allergic alveolitis (EAA) is the paradigm of lung response to inhaled organic material, either in the form of particles or of organic protein, or even of simple chemicals, organic and inorganic. Given its etiologic multiplicity and diversity of clinical, HP has been considered as a syndrome, with highly variable and difficult to assess prevalence.
In a population study, the estimated annual incidence of interstitial lung diseases was 30 per 100,000 individuals [1, 2]. In that study, the HP was less than 2% of cases.
Several diagnostic criteria for HP proposals have been published, the most used is by Richerson et al [1, 3].
Clinically, can be considered three forms of presentation - acute, subacute and chronic - depending on the nature of the aggressor agents, their physical characteristics, their concentration, the intensity of individual exposures and individual variability. The acute response after inhalation is a nonspecific diffuse pneumonitis with inflammatory cell infiltration of the bronchioles, alveoli, and interstitium. In the subacute and chronic stages, loosely formed, noncaseating, epithelioid cell granulomas may be dispersed in the interstitium.
Given a picture of a single determinant HP, its eviction is mandatory. The treatment is based on corticotherapy, despite the absence of long-term benefit evidence [4].
| Case Report | ▴Top |
The authors present the case of a patient 50 years old black male, born in Angola and a resident of Cantanhede, Portugal, former factory worker (he worked during 5 years in a metallurgical manufactures, with lead exposure, until 16 years ago), observed at the Emergency Room (ER) of the Hospitais da Universidade de Coimbra (HUC) in November 2007, with complaints of weakness, easy fatigability for moderate exertion, exertional dyspnea, nonproductive cough, which had been steadily evolving for six months, still referring not quantified weight loss.
He had no family or personal history of pathological relevance. He denied smoking and related consumption of approximately 50 g of alcohol per day. He was not on any kind of medication and epidemiological history was irrelevant.
On physical examination the patient was alert, oriented and collaborative temporo-spatially, flushed and hydrated, afebrile with a respiratory rate of 20 cpm, blood pressure 135/75 mm Hg and pulse oximetry 95% on room air. The pulmonary auscultation revealed a slight overall decrease of vesicular murmur and bilateral crackles. The cardiac auscultation and abdominal examination showed no changes. He had no palpable lymphadenopathy or peripheral edema, and the remaining physical examination was normal.
The patient had some complementary diagnostic exams performed on an outpatient basis during the previous six months. Of these tests we highlight the elevation of serum angiotensin converting enzyme (SACE) (146 U/L) and erythrocyte sedimentation rate (ESR) (77 mm/h), as well as an opacity of a fibrotic appearance in both lung fields on chest X-ray, although most exuberant in the middle level of the right lung field (Fig. 1).
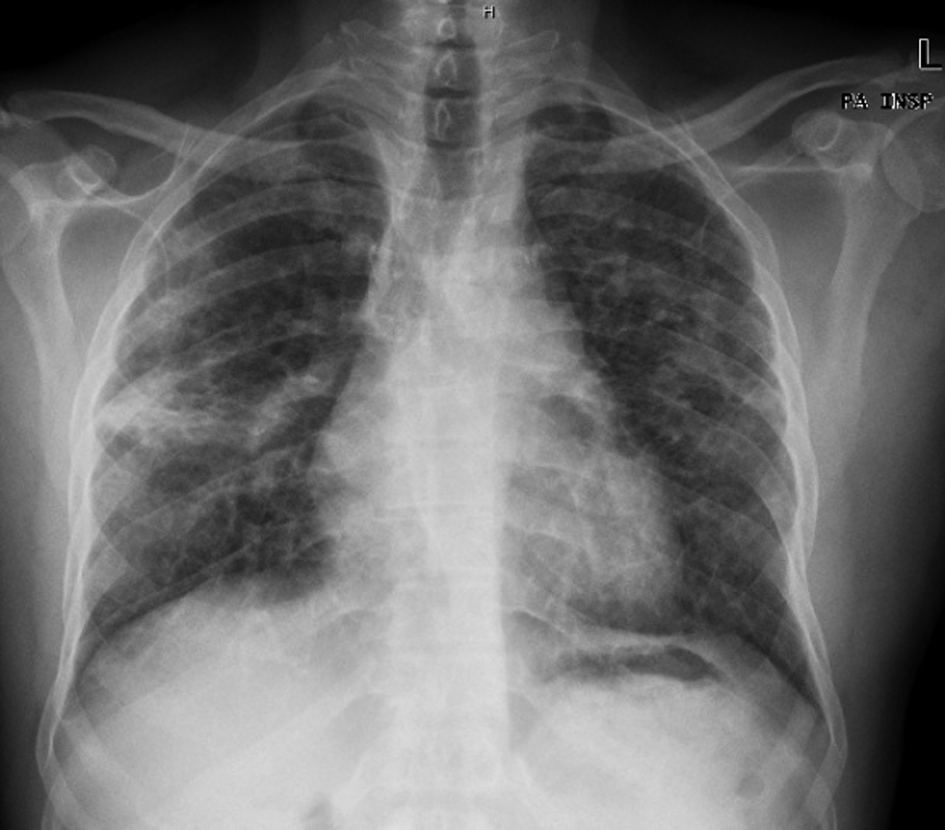 Click for large image | Figure 1. Chest X-ray showing opacity of a fibrotic appearance in both lung fields, most exuberant in the middle level of the right lung field. |
The chest CT revealed areas of fibrosis and thickening of interlobular septa in both lung bases and at the apical segments of upper lobes, some areas of localized densification in ground glass appearance in both lower lobes and multiple enlarged mediastinal lymph nodes (Fig. 2).
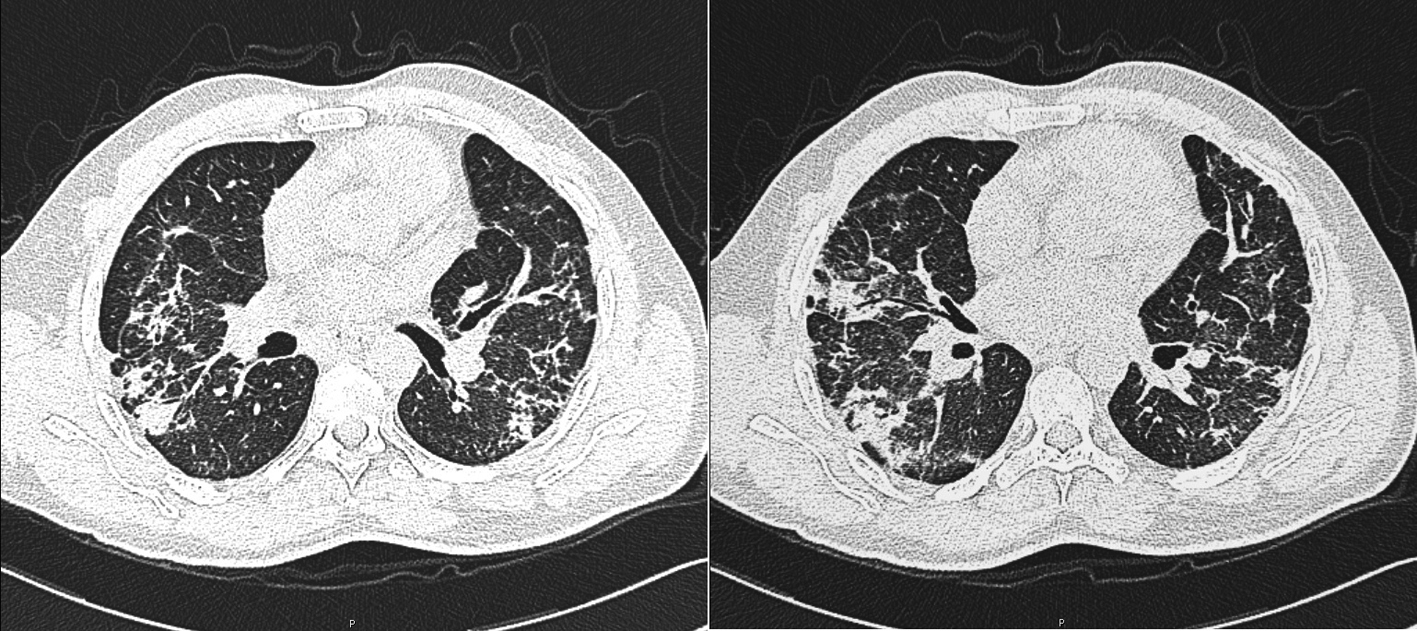 Click for large image | Figure 2. Chest CT showing areas of fibrosis and thickening of interlobular septa in both lung bases. |
The imaging tests identified lung fibrosis. The clinical setting associated with it leads us to think this as a disease associated with fibrosis: a) an occupational disease causing interstitial pneumonitis (extrinsic allergic asthma), b) infectious disease, including pulmonary tuberculosis, c) sarcoidosis or systemic autoimmune disease, d) idiopathic pulmonary fibrosis.
In this context, the patient was then hospitalized at the Internal Medicine Department for further study.
From the exams carried out during hospitalization we highlight a hypoxemia of 65.3 mm Hg in arterial blood gas analysis. The electrophoretic protein profile revealed an IgA Kapa monoclonal gammopathy. The 24-h urine calcium was normal (216 mg/time). HIV and Mantoux tests were negative. Blood lead determination was also performed, which proved normal: 75 mg/L (reference range: 40 - 290 mg/L). The optic bronchoscopy and bronchoalveolar lavage (BAL) showed no changes had a CD4/CD8 ratio of 3.12. Respiratory function tests (RFT) showed a severe restrictive syndrome with decreased severe alveolocapillary diffusion by carbon monoxide. The gallium scintigram showed intense abnormal tracer uptake throughout the lung parenchyma, bilaterally as well as the ganglionic level (Fig. 3).
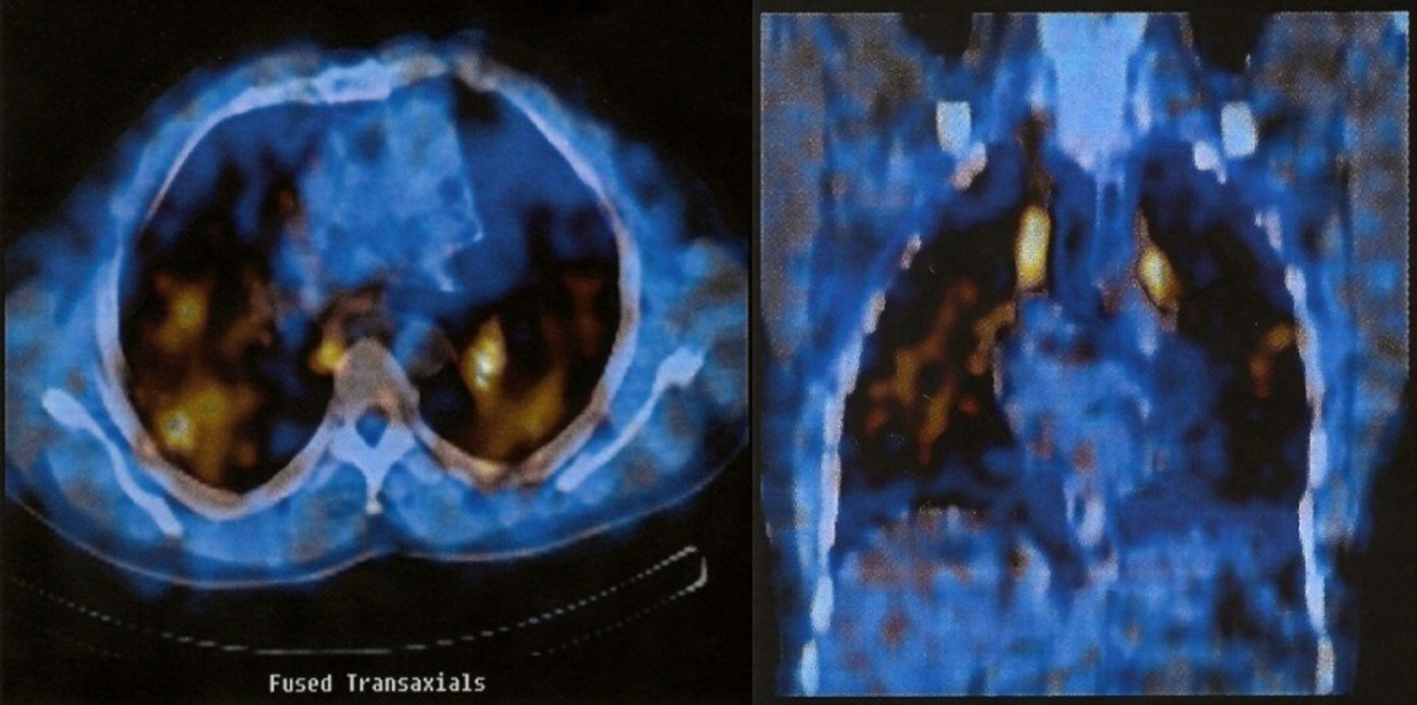 Click for large image | Figure 3. Gallium scintigram showing intense abnormal tracer uptake throughout the lung parenchyma, bilaterally as well as the ganglionic level. |
Lymph node biopsy was performed by mediastinoscopy, whose histology revealed exuberant histiocytic hyperplasia and sinus histiocytosis with epithelioid granulomas of mediastinal lymph nodes, all of small dimension.
Cultures of biological products, including BAL, bronchial aspirates, blood and urine were negative, so as antinuclear antibodies, ANCA and anti-glomerular basement membrane antibodies.
At this stage it seemed as most likely diagnosis the occupational interstitial lung disease (chronic granulomatous disease of unknown etiology professional, including hypersensitivity pneumonitis) but the possibility of sarcoidosis could not definitively excluded, since he didn’t have contact with his professional environment for some time and there was no significant improvement.
During hospitalization was treated with prednisolone 1 mg/kg/day with clinical improvement and the patient was discharged to the outpatient service, keeping corticotherapy (20 mg prednisolone/day).
The situation remained stable for about eight months, but in May 2008, due to worsening respiratory symptoms (respiratory failure), was again hospitalized. As there wasn’t a definitive diagnosis, it was decided to perform transthoracic lung biopsy. This revealed abnormalities suggestive of unspecified interstitial pneumonitis. We chose to keep the patient on corticotherapy (40 mg prednisolone/day), and initiated long-term oxygen therapy.
In September 2008, a respiratory infection conditioned clinical worsening with severe respiratory failure and need for assisted ventilation, being admitted to the Intensive Care Unit (UCI). Given the inconclusive results of previous histology and the absence of clinical and radiological response to therapy instituted during this hospitalization, an open lung biopsy was performed, which revealed alveolar distension, constrictive bronchiolitis and bronchitis, due to the smooth muscle cell hyperplasia in the wall of the terminal and respiratory bronchioles. In these areas of alveolar distension, the inter-alveolar septa were thin and there were no macrophages. Along the bronchovascular axis there were epithelioid granulomas with rare multinucleated giant cell with cytoplasm pigmentation as well as cholesterol crystals. Focal loss of lobular structure was due to pulmonary confluence of non-necrotizing epithelioid granulomas without giant multinucleated Langhans cells, in response to phagocytosis of lipid products (cytoplasm pigmentation and cholesterol crystals). There was no regular distribution of epithelioid granulomas, which apparently evolved along the bronchovascular axis, converging in peripheral subpleural regions. There wasn’t BALT hyperplasia or specific inflammatory infiltrate. Thus was made the histopathological diagnosis of bronchiolitis and hypersensitivity pneumonitis by inhalation lipid (compatible with inhalation-related products and lead paint) (Fig. 4).
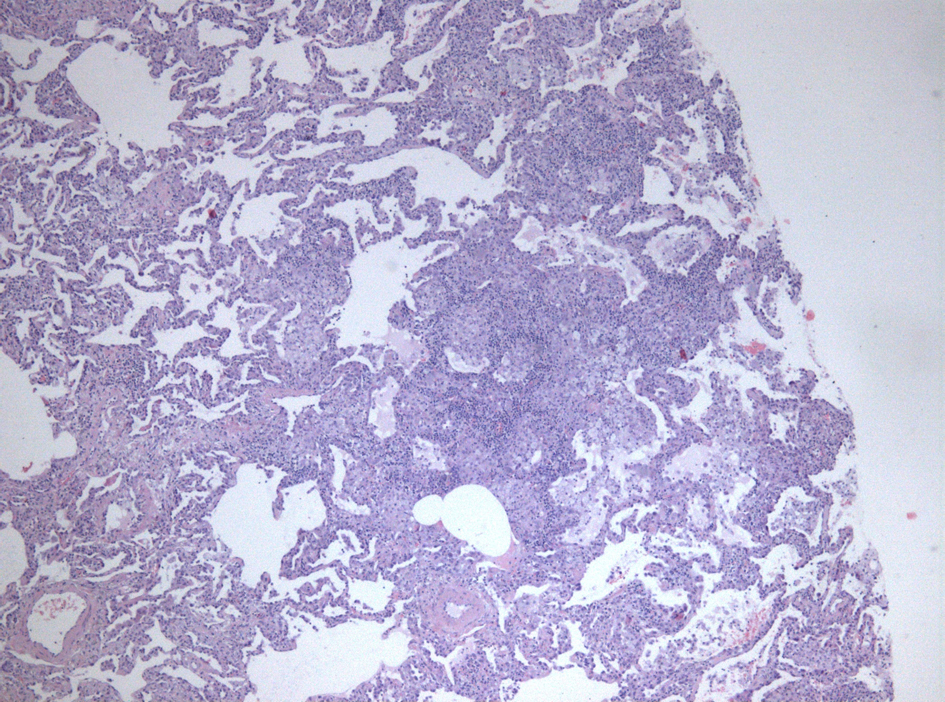 Click for large image | Figure 4. Open lung biopsy, 200 × magnification, 2008. |
The combination of azathioprine (100 mg/day) with corticotherapy allowed clinical improvement.
During 2009, the patient had several hospitalizations for infectious complications with worsening respiratory failure. In December of that year he was again admitted to the ICU with community-acquired pneumonia with respiratory failure and need for assisted ventilation. He died on January 9, 2010 due to nosocomial pneumonia by multiresistant Acinetobacter baumannii.
| Discussion | ▴Top |
Hypersensitivity pneumonitis, also known as extrinsic allergic alveolitis, is a syndrome caused by repeated inhalation of specific antigens from occupational or environmental exposure in sensitized individuals. Hypersensitivity pneumonitis is considered a granulomatous interstitial disease of the lungs [5].
The first cases of HP were described at the beginning of the twentieth century, in farmers exposed to hay or straw. Since then it has been attributed to inhalation of various antigens found in the environment [1]. HP was first reported among metal machining workers in 1995. In the following years, additional outbreaks were reported [6, 7]. The most recent outbreak was reported in 2007 from an engine manufacturing facility in England [8].
HP results from an exaggerated immune response, leading to the onset of symptoms similar to an acute or progressive lung damage, sometimes irreversible [1], as with our patient.
The clinical features and severity of symptoms vary according to the frequency and intensity of exposure. A history of exposure to potential agents or changes in the domestic and other environments (or both) is essential to diagnosis and treatment. The interval between exposure to the antigen and clinical manifestations of lung disease is unknown, although symptoms can occur as soon as 4 - 12 h after exposure. In more chronic and low-level exposures, however, the onset is insidious. Our case represents the lack of specificity of the complaints and the long evolution of symptoms until the patient sought health care.
Further avoidance of exposure to the antigen or antigens and treatment with corticosteroids are important if improvement is to be obtained. Continued exposure to the unidentifiable antigens or prolonged exposure to antigens, or both, have led to chronic hypersensitivity pneumonitis and irreversible fibrosis that may not respond to any treatment regimen. The resistance to corticotherapy and the modest response with the association of other immunosuppressants such as azathioprine, was evident in the case of our patient.
PH is an underdiagnosed entity; therefore its exact prevalence is unknown. In addition, the diagnosis is sometimes particularly difficult, since the clinical picture is not specific. In our case it was not possible to identify the patient’s past any acute episode suggestive of this type of pathology, particularly during the period that he had occupational exposure to the allergen in question. However, there is reference to some patients in whom prolonged and continuous exposure to small amounts of antigen resulted in irreversible lung damage without acute illness [9] .In these situations, pulmonary fibrosis becomes prominent and only in advanced stages [10] arise progressive dyspnea, coughing, malaise, anorexia and significant weight loss, without fever, with bibasal crackles and no digital clubbing, as in our patient.
Also relevant in this case report was the necessity to do three biopsies (lymph node and lung). The first (lymph node biopsy) was done in the diagnostic approach in the search of the causes of pulmonary fibrosis, since the exams, including nuclear imaging and BAL were inconclusive. Pathology did not allow any significant progress in relation to the clinical diagnoses proposed, namely, occupational interstitial lung disease and sarcoidosis. The second biopsy (transthoracic lung) was performed because there was no established diagnosis and patient was not improving with corticotherapy. The anatomo-pathological examination revealed abnormalities suggestive of unspecified interstitial pneumonitis. Corticotherapy was kept, with long-term oxygen therapy at home, due to worsening respiratory failure. Since the results of this biopsy were not conclusive and progressive worsening occurred, in September 2008 an open lung biopsy was performed. Pathologic examination allowed the diagnosis of bronchiolitis and hypersensitivity pneumonitis by lipid inhalation, consistent with the inhalation of paint-related products and lead. It was therefore decided to associate azathioprine (100 mg/day) to the corticotherapy, with frank improvement in clinical status.
Knowledge of each HP evolution is essential, particularly in the professional level, with legal and economic implications. However, it is not easily predictable, depending on several factors, from the nature and intensity of aggression to the characteristics of each individual.
The prognosis of chronic HP largely depends upon the extent of permanent lung damage at the time of diagnosis [11]. The small percentage of patients who develop advanced pulmonary fibrosis may have continued progression of their disease, even though they completely avoid re-exposure to the inciting antigen [11, 12]. Such patients may develop severe, chronic hypoxemia and require continuous oxygen therapy [11], as in our patient.
Although there are no controlled clinical trials regarding the treatment of chronic HP, it is recommended that patients with severe or progressive chronic HP be treated with trial corticosteroids [11, 13].
Lipid inhalation HP is rare and the lung toxicity associated with it is serious and potentially fatal, as evidenced by the case presented. The authors present this case not only for its rarity but also for the initial histopathological feature simulating sarcoidosis.
Acknowledgments
Our thanks to Professor Lina Carvalho, from the Pathology Department of the Hospitais da Universidade de Coimbra, for the cooperation in the diagnosis and for the histologic image.
Conflict of Interest
None.
| References | ▴Top |
- Girard M, Lacasse Y, Cormier Y. Hypersensitivity pneumonitis. Allergy. 2009;64(3):322-334.
pubmed doi - Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150(4):967-972.
pubmed - Richerson HB, Bernstein IL, Fink JN, Hunninghake GW, Novey HS, Reed CE, Salvaggio JE, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844.
pubmed doi - Monkare S, Haahtela T. Farmer's lung—a 5-year follow-up of eighty-six patients. Clin Allergy. 1987;17(2):143-151.
pubmed doi - De Vuyst P, Dalphin JC. [Occupational interstitial lung diseases]. Rev Prat. 2007;57(20):2266-2276.
pubmed - Gupta A, Rosenman KD. Hypersensitivity pneumonitis due to metal working fluids: Sporadic or under reported? Am J Ind Med. 2006;49(6):423-433.
pubmed doi - Hodgson MJ, Bracker A, Yang C, Storey E, Jarvis BJ, Milton D, Lummus Z, et al. Hypersensitivity pneumonitis in a metal-working environment. Am J Ind Med. 2001;39(6):616-628.
pubmed doi - Rosenman KD. Asthma, hypersensitivity pneumonitis and other respiratory diseases caused by metalworking fluids. Curr Opin Allergy Clin Immunol. 2009;9(2):97-102.
pubmed doi - N. Franklin Adkinson, JR e col. Allergy – Principles & Practice – 6a Edicao – Mosby 2007.
- Jason S Voulekis e col. The effect of Pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. The American Journal of Medicine – Maio 2004.
- Lawrence CM. Hypersensitivity pneumonitis. Curr Opin Pulm Med 2004;10:401-411.
- Patel AM, Ryu JH, Reed CE. Hypersensitivity pneumonitis: current concepts and future questions. J Allergy Clin Immunol. 2001;108(5):661-670.
pubmed doi - Bertorelli G, Bocchino V, Olivieri D. Hypersensitivity pneumonitis - In Interstitial Lung Diseases. European Respiratory Monograph 2000;14:120-136.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.