

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 3, Number 2, April 2012, pages 149-152
Familial Balanced Reciprocal Translocation [t(16;22)(p11;q13)mat] in a Child With Constitutional Short Stature
Halit Akbasa, d, Orhan Koksalb, Capan Koncab, Mahmut Balkanc, Turgay Budakc
aDepartment of Medical Biology, Medical Faculty, University of Harran, 63100 Sanllurfa, Turkey
bDepartment of Pediatrics, Medical Faculty, University of Dicle, 21100 Diyarbaklr, Turkey
cDepartment of Medical Biology and Genetics, Medical Faculty, University of Dicle, 21100 Diyarbaklr, Turkey
dCorresponding author: Halit Akbas, Department of Medical Biology, Medical Faculty, University of Harran, 63100 Sanllurfa, Turkey
Manuscript accepted for publication January 27, 2012
Short title: Familial Balanced Reciprocal Translocation
doi: https://doi.org/10.4021/jmc534w
| Abstract | ▴Top |
One of the reasons of most frequent application to Clinics of Pediatric Endocrinology is short stature. The major reasons are familial short stature, constitutional delay, systematic diseases, lack of nutrition and endocrine disorders. Chromosomal analysis is required for cases whose reason cannot be detected and especially has a dysmorphic appearance. Our patient was a 12 year old boy who had applied due to complaint of short stature, with no detected pathology in routine examinations and in whom reciprocal translocation that scoped chromosomes number 16 and 22 in the cytogenetic analysis conducted in the Genetic Diagnosis Laboratory was detected. In the cytogenetic examination conducted on family members, it has been detected that the child’s mother and mother’s father carried the same translocation, however, did not have any visible physical anomalies. According to the literature data at hand, our patient is the first short stature case that is a carrier of balanced reciprocal translocation between the 16 and 22 numbered chromosomes. In this study, the patient’s clinical and laboratory characteristics, detailed family history and genetic study results are discussed under the light of the literature information.
Keywords: Chromosome; Balanced translocation; Short stature
| Introduction | ▴Top |
Chromosomal anomalies are divided into two as numerical and structural. Translocations are the most frequently encountered in structural anomalies at 1/500 frequency. Translocations are divided into three sub-groups as reciprocal, centric fusion (Robertsonian) and insertional. Reciprocal translocations are formed by means of breakage in non-homolog chromosomes and the mutual change of location of the broken fragments. If after the translocation there is no loss of the fragments transmitted due to translocation or if a fragment is not added to the genome as an extra, this is called a balanced translocation. Reciprocal translocations do not mostly give phenotypic findings in event that they are balanced. However, phenotypic influence can be seen in the children of these individuals, subject to the partial monosomy or partial trisomy in the relevant chromosomes [1].
One of the reasons of most frequent application to Polyclinics of Pediatric Endocrinology is short stature. The major reasons scope familial short stature, constitutional delay, systematic diseases, lack of nutrition and endocrine disorders [2]. Chromosomal examination is required for cases whose reason cannot be detected and especially has a dysmorphic appearance.
In this study, a case which was applied to the Polyclinics of Pediatric Endocrinology for due to growth and development delay in which balanced reciprocal translocation scoping the 16 and 22 numbered chromosomes in a cytogenetic analysis conducted in the Genetic Diagnosis Laboratory was detected.
| Case Report | ▴Top |
Our patient was a 12-year-old boy who had applied to the Polyclinics of Pediatric Endocrinology due to underdevelopment and short stature which was observed in four years. It was seen in the detailed family history that there is no member having a similar complaint. The patient’s, who has a neuromotor development in accordance with his peers, body weight was measured as 23 kg (< 3% percentile) and height as 124 cm (< 3% percentile). The bone age of the patient, who was assessed as prepubertal, was determined as 9. The hemogram, liver and kidney functionalty tests, serum electrolytes, urine and stool examinations in the laboratory examinations were normal. No significant finding was observed during the examinations for endocrine, infection and malabsorption.
A genetic examination was requested to determine the reason of the short stature. It was determined that the patient had a 46, XY, t(16;22)(p11;q13) chromosome constitution (Figs. 1, 2). The pedigree of the patient was issued and a chromosome analysis was performed his relatives, who could be reached, so as to determine the origin of the reciprocal translocation. It was determined that the patient’s mother (II-7) and the mother’s father (I-3) had the same chromosome constitution and the father, brother and sister had a normal karyotype (Fig. 3). With the available findings, the patient was diagnosed with constitutional short stature.
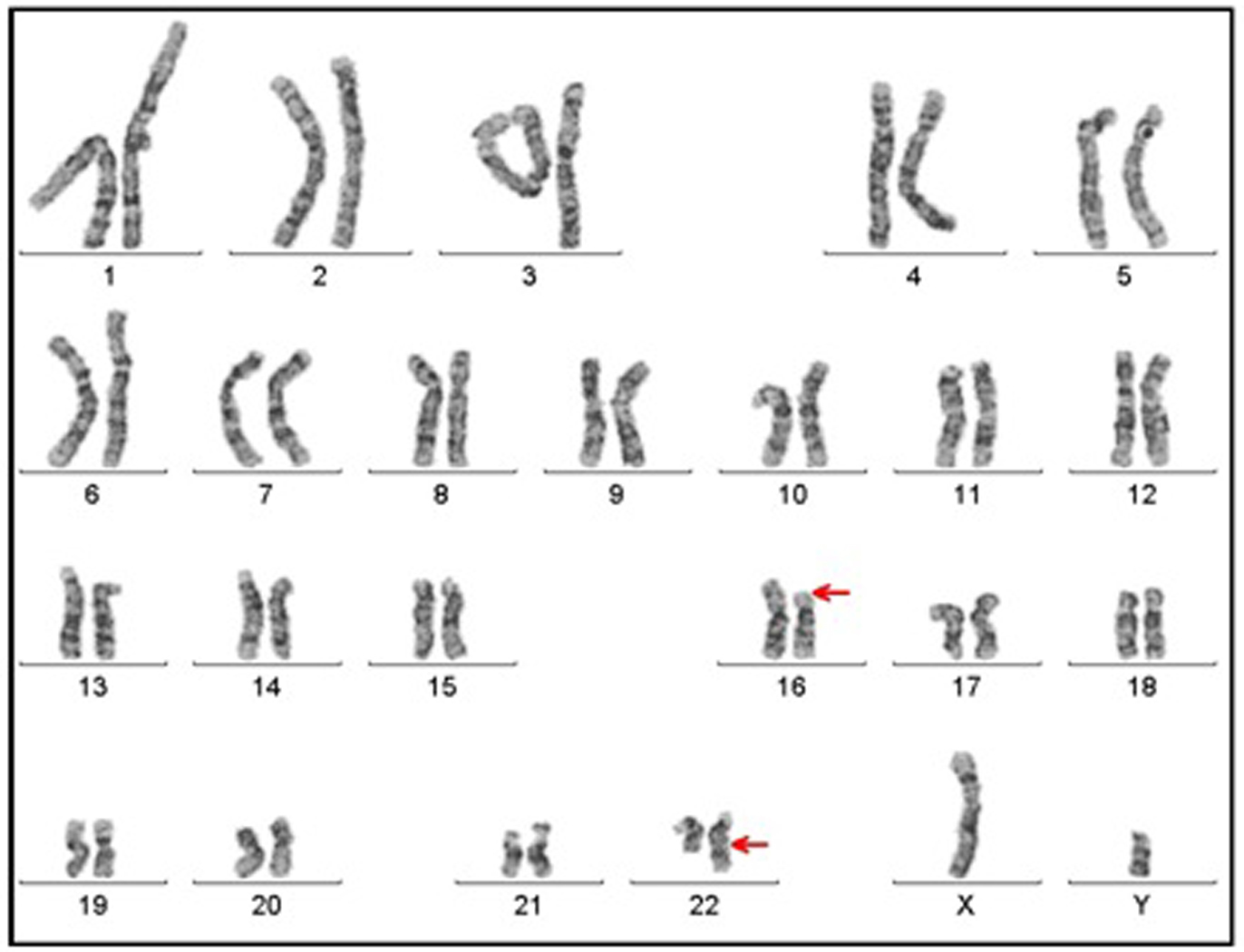 Click for large image | Figure 1. Karyotype having 46, XY, t(16;22)(p11;q13) chromosome constitution (arrows show breakpoints). |
 Click for large image | Figure 2. Partial karyotype of case having 46, XY, t(16;22)(p11;q13) chromosome constitution (breakpoints have been indicated on the ideogram). |
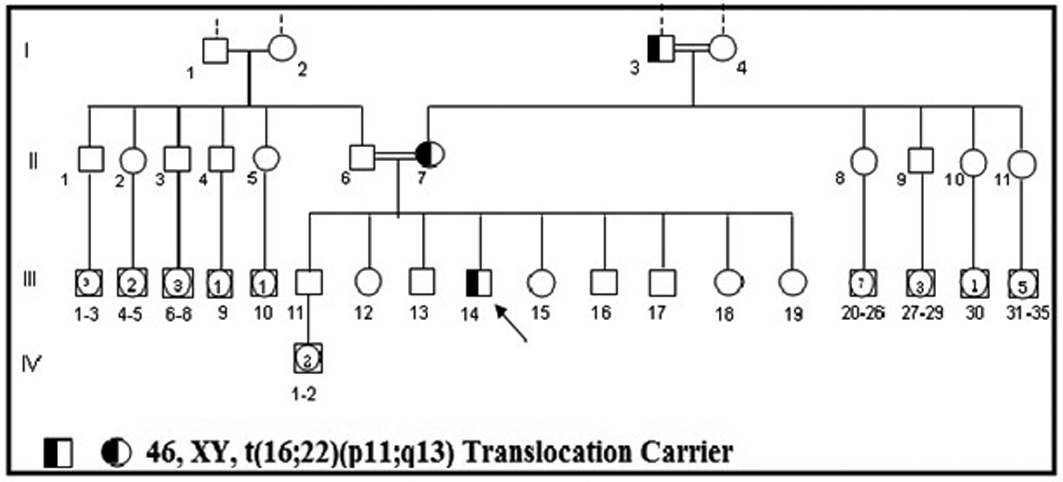 Click for large image | Figure 3. Family pedigree of the case carrying 46, XY, t(16;22)(p11;q13) translocation points. |
Peripheral blood was taken under sterile circumstanced with a heparinised injector from each case for the chromosome analysis and the whole blood technique, which is the modified technique of the macro-culture technique developed by Moorhead et al. was applied [3, 4]. The preparations were stained with the Giemsa Banding Technique (GTG) and the chromosomes in the 30 - 50 metaphase plate was evaluated in terms of the numerical and structural anomalies and a karyotype was made from 25 banded metaphases. The cytogenetic analysis results were reported according to ISCN 1995 [5].
| Discussion | ▴Top |
It is expected that the breakages leading to the formation of balanced translocations do not cause great piece loss in the chromosomes, consequently, that there will not much gene loss in the person and no change will occur in the phenotype of the person. Nevertheless, apparently balanced translocations form a widespread type of chromosomal anomalies that can be seen both in patients that have an abnormal phenotype and healthy individuals that have not been influenced. Approximately one fifth of these anomalies, which are mostly familial, is de novo [6]. The risk of finding abnormal phenotypes in individuals having de novo regulation is approximately 6.7% [7]. There are balanced translocations holding different chromosomes associated with short stature or development delay are available in literature [8, 9]. Cytogenetically balanced reciprocal translocation is also observed in our case that has short stature exists. According to the literature data at hand, this case is the first balanced reciprocal translocation case scoping 16 and 22 chromosomes with structural short stature.
Finding the same translocation in our case’s mother and mother’s father, who have normal phenotypes, however both of them having a normal height during their childhood ages, makes our patient even more interesting. Along with complying with structural short stature with the available findings, we wonder what the final height will be.
Even under the best of circumstances, changes under 4 kb (kilobasis) cannot be analysed with chromosome analysis [10]. The familial balanced translocation’s phenotypic affects can change due to imbalanced de novo regulations on a molecular level, uniparental disomy associated with translocationed chromosomes, disruption of genes located in the breakpoints, translocation indicating mosaicism in another tissue or modification of expression of the gene carried somewhere else in the genome [11-13]. Hence, Baptista et al. [14] have stated that as a result of the study in which they have conducted the molecular cytogenetic analysis of breakpoints in balanced reciprocal translocation carriers by means of fluorescence in situ hybridization (FISH) and array CGH methods, balanced reciprocal translocations do not bear imbalance in breakpoints in phenotypically normal patients and that the breakpoints in the translocations of patients, who are phenotypically abnormal, is mainly associated with cryptic imbalances. In addition to this, the mutations in the SHOX (Short Stature Homeobox) gene located in the pseudoaotomosal regions of the X and Y chromosomes’ short arms are held responsible of idiopathic short stature [15, 16]. In our case, a mutation that has occurred in the SHOX gene independently from reciprocal translocation may have also led too short stature. It was planned to research the translocation on a molecular level in our patient, however, the family has refused to this request to do socio-economic reasons. The answer to whether or not the translocation has left damage on a molecular level has not been found, however, the patient has been taken under periodic monitoring.
As a result, balanced reciprocal translocation may appear under a specific part of structural short statures. However, in order to give a healthy genetic counseling to individuals who have abnormal phenotypes such as short stature in which balanced reciprocal translocation is detected in the chromosome analysis, advanced molecular cytogenetic and molecular genetic methods should be referred to especially for the breakpoints regions.
| References | ▴Top |
- Başaran N. Medical Genetics. 7.th ed. Nobel Tıp Publishing Ltd. 1999: Chp. 9:161-163.
- Ratel L., Cloyton PE. Normal and disordered growth. In: Brook’s Clinical Pediatric Endocrinology: 5th ed. Blackwell Puplishing Ltd 2005:101-103.1.
- Balkan M, Alp MN, Budak T. A Balanced Reciprocal Translocation Case in Family with a History of Recurrent Abortions. Dicle Medical Journal 35(1): 61-64.
- Moorhead PS, Nowell PC, Mellman WJ, Battips DM, Hungerford DA. Chromosome preparations of leukocytes cultured from human peripheral blood. Exp Cell Res. 1960;20:613-616.
pubmed - Shaffer LG, Tommerup N. ISCN-An international system for human cytogenetic nomenclature, 2005.
- Jacobs PA, Browne C, Gregson N, Joyce C, White H. Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J Med Genet. 1992;29(2):103-108.
pubmed doi - Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991;49(5):995-1013.
pubmed - Rao VB, Lily K, Seema K, Ghosh K, Dipika M. Paternal reciprocal translocation t(11;16)(p13;q24.3) in a Silver-Russel syndrome patient. Ann Genet. 2003;46(4):475-478.
pubmed doi - Rao L, Kanakavalli M, Padmalatha V, Nallari P, Singh L. Paternally derived translocation t(8;18)(q22.1;q22)pat associated in a patient with developmental delay: Case report and review. J Pediatr Neurosci. 2010;5(1):64-67.
pubmed doi - Gardner McKinlay RJ, Sutherland GR. Chromosome Abnormalities and Genetic Counseling. 3rd Edition, Oxford Un. Press. UK. 2004.
- Tonk VS, Wyandt HE, Huang X, Patel N, Morgan DL, Kukolich M, Lockhart LH, et al. Disease associated balanced chromosome rearrangements (DBCR): report of two new cases. Ann Genet. 2003;46(1):37-43.
pubmed doi - A. Schinzel, Catalogue of Unbalanced Chromosome Aberrations in Man,Walter de Gruyter, NewYork, 2001 pp. 20.
- Kumar A, Becker LA, Depinet TW, Haren JM, Kurtz CL, Robin NH, Cassidy SB, et al. Molecular characterization and delineation of subtle deletions in de novo "balanced" chromosomal rearrangements. Hum Genet. 1998;103(2):173-178.
pubmed doi - Baptista J, Prigmore E, Gribble SM, Jacobs PA, Carter NP, Crolla JA. Molecular cytogenetic analyses of breakpoints in apparently balanced reciprocal translocations carried by phenotypically normal individuals. Eur J Hum Genet. 2005;13(11):1205-1212.
pubmed doi - Zinn AR, Wei F, Zhang L, Elder FF, Scott CI, Jr., Marttila P, Ross JL. Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet. 2002;110(2):158-163.
pubmed doi - Rappold GA, Fukami M, Niesler B, Schiller S, Zumkeller W, Bettendorf M, Heinrich U, et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab. 2002;87(3):1402-1406.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.