

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 14, Number 12, December 2023, pages 405-412

Idiopathic Hypertrophic Spinal Pachymeningitis
Ashutosh Guptaa, Daniel Uma, Rohan Samantb, Rodrigo Hasbunc, Rohini D. Samudralward, Shitiz Sriwastavae, Rajesh K. Guptae, f
aMcGovern Medical School, UT Health Science Center at Houston, Houston, TX 77030, USA
bDepartment of Radiology, UT Health Science Center at Houston, TX 77030, USA
cDepartment of Infectious Disease, UT Health Science Center at Houston, TX 77030, USA
dDepartment of Neurology, University of Pennsylvania, Philadelphia, PA, USA
eDivision of Multiple Sclerosis and Neuroimmunology, Department of Neurology, University of Texas Health Science Center at Houston, Houston, TX 77030, USA
fCorresponding Author: Rajesh K. Gupta, Division of Multiple Sclerosis and Neuroimmunology, Department of Neurology, University of Texas Health Science Center at Houston, Houston, TX 77030, USA
Manuscript submitted August 23, 2023, accepted November 2, 2023, published online December 29, 2023
Short title: Idiopathic Hypertrophic Spinal Pachymeningitis
doi: https://doi.org/10.14740/jmc4149
| Abstract | ▴Top |
Hypertrophic pachymeningitis (HP) is a rare presentation with duramater thickening and fibrosis which can result in cranial or spinal compressive disease. Most cases of spinal HP require surgical management. We present an uncommon case of idiopathic hypertrophic spinal pachymeningitis (IHSP) in a 40-year-old male who showed complete improvement to steroids without any further relapses. The patient presented with bilateral upper limb weakness with magnetic resonance imaging (MRI) spine showing diffuse dural thickening of the entire spine with cervical cord compression. He had an extensive workup for underlying etiology and worsening symptoms until he was diagnosed with IHSP. Later, he was started on high-dose steroids with good response and no relapse after 2 years. A descriptive analysis of IHSP cases since 2009 including ours showed that it usually occurs after 50s with female preponderance. Weakness and sensory loss are the most common complaints with 50% patients showing clinical signs of myelopathy like hyperreflexia, clonus, Babinski sign and sensory level. Cerebrospinal fluid (CSF) and inflammatory markers like erythrocytic sedimentation rate (ESR) and C-reactive protein (CRP) can be used to assess disease progression and prognosis. Surgical removal of HP followed by steroids is the best line of management while steroids alone can be tried in cases where clinical signs of myelopathy are absent.
Keywords: IHSP; Hypertrophic pachymeningitis; Idiopathic; Steroids; Spinal cord compression
| Introduction | ▴Top |
Hypertrophic pachymeningitis (HP) is an uncommon inflammatory disorder causing chronic diffuse fibrosis and thickening of duramater [1, 2]. HP can be classified into primary or idiopathic cases and secondary cases when an underlying etiology is identified. The common etiologies for secondary cases include infection, tumor, neurosarcoidosis, granulomatosis with polyangiitis, and IgG4-related disease. The underlying pathophysiology of idiopathic HP is not well understood and there are multiple hypotheses regarding the mechanism of the disease. One hypothesis is that it is an autoimmune disorder arising from infiltrative or infectious etiologies. Biopsies of duramater in patients with HP have shown fibrosis and CD4+ T-cell inflammatory infiltrates [2, 3].
HP can occur anywhere in the craniospinal axis from the brain to spinal cord. Spinal form of HP usually presents with weakness, ataxia and sensory loss due to compression of surrounding structures and is one of the rare causes of radiculomyelopathy. Most cases with spinal HP require surgical decompression to prevent neurological complications and relapses [4]. Here, we present a case of idiopathic hypertrophic spinal pachymeningitis (IHSP) responding to steroids with no relapse. We also provide a review of current literature of all IHSP cases since 2009, focusing on their clinical spectrum, diagnosis and management.
| Case Report | ▴Top |
Investigations
A 40-year-old man with ankylosing spondylitis presented to our clinic with a 4-week history of progressively worsening bilateral upper limb weakness. He had a past history of spinal tuberculosis (TB) causing bilateral arm paresthesia for 9 years after extensive workup and treated with standard TB regimen. He complained of difficulty in wearing clothes and taking showers due to his arm weakness. The patient does not experience falls but exhibits noticeable limp while walking and occasional episode of ataxia. He denied any myalgia, fasciculations, cramping or atrophy. There is no history of headache, seizures or any other neurological complain. On neurological examination, he had mild weakness of bilateral upper extremities (power 4/5) proximally and distally, rest non-significant. He was unable to abduct his arms greater than 90°. Magnetic resonance imaging (MRI) of the brain showed enhanced dural thickening in the posterior fossa (Fig. 1a, b). Additionally, an incidental right cerebellopontine angle mass was observed in his MRI of brain, although it was considered unlikely to be contributing to his symptoms due to their location (Fig. 1b, c).
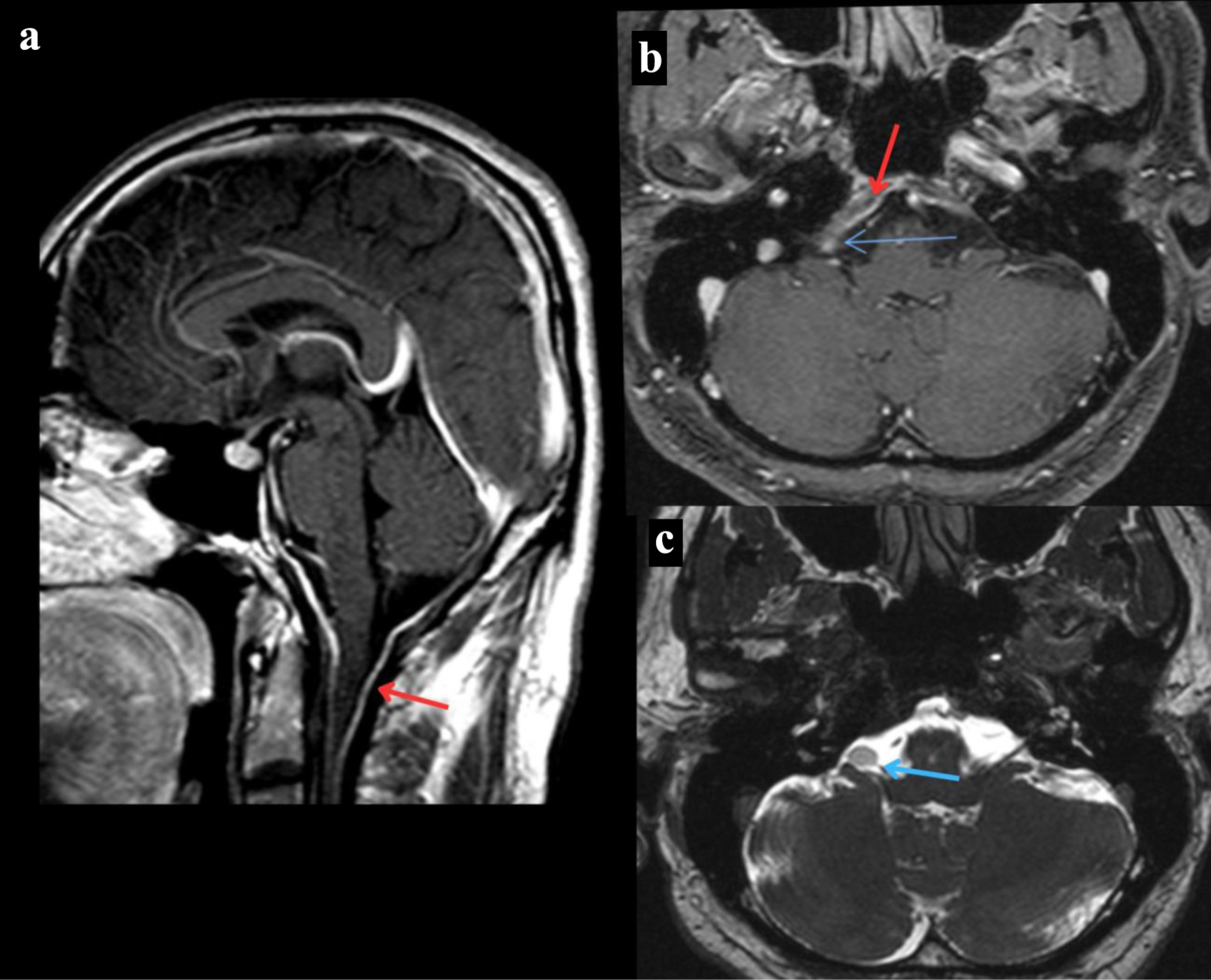 Click for large image | Figure 1. MRI of the brain. (a) T1-weighted MRI of the brain sagittal section post-contrast that reveals dural thickening of the posterior fossa indicated by the red arrow. (b) T1-weighted MRI of the brain axial section post-contrast that reveals a right cerebellopontine angle mass revealed by the blue arrow and dural thickening of the posterior fossa indicated by the red arrow. (c) T2-weighted MRI of the brain axial section that reveals a right cerebellopontine angle mass revealed by the blue arrow. MRI: magnetic resonance imaging. |
MRI of the spine showed diffuse dural thickening of the entire spine with compression at the cervical spinal cord from C2 level to the upper thoracic spine T2 level (Fig. 2a-d). MRI of the lumbar spine showed grade 1 anterolisthesis at the level of L5-S1 secondary to bilateral pars defect with moderate foraminal stenosis at L5-S1. In addition, moderate to advanced multilevel facet arthropathy was shown at levels L2-S1, producing moderate foraminal stenosis at multiple levels.
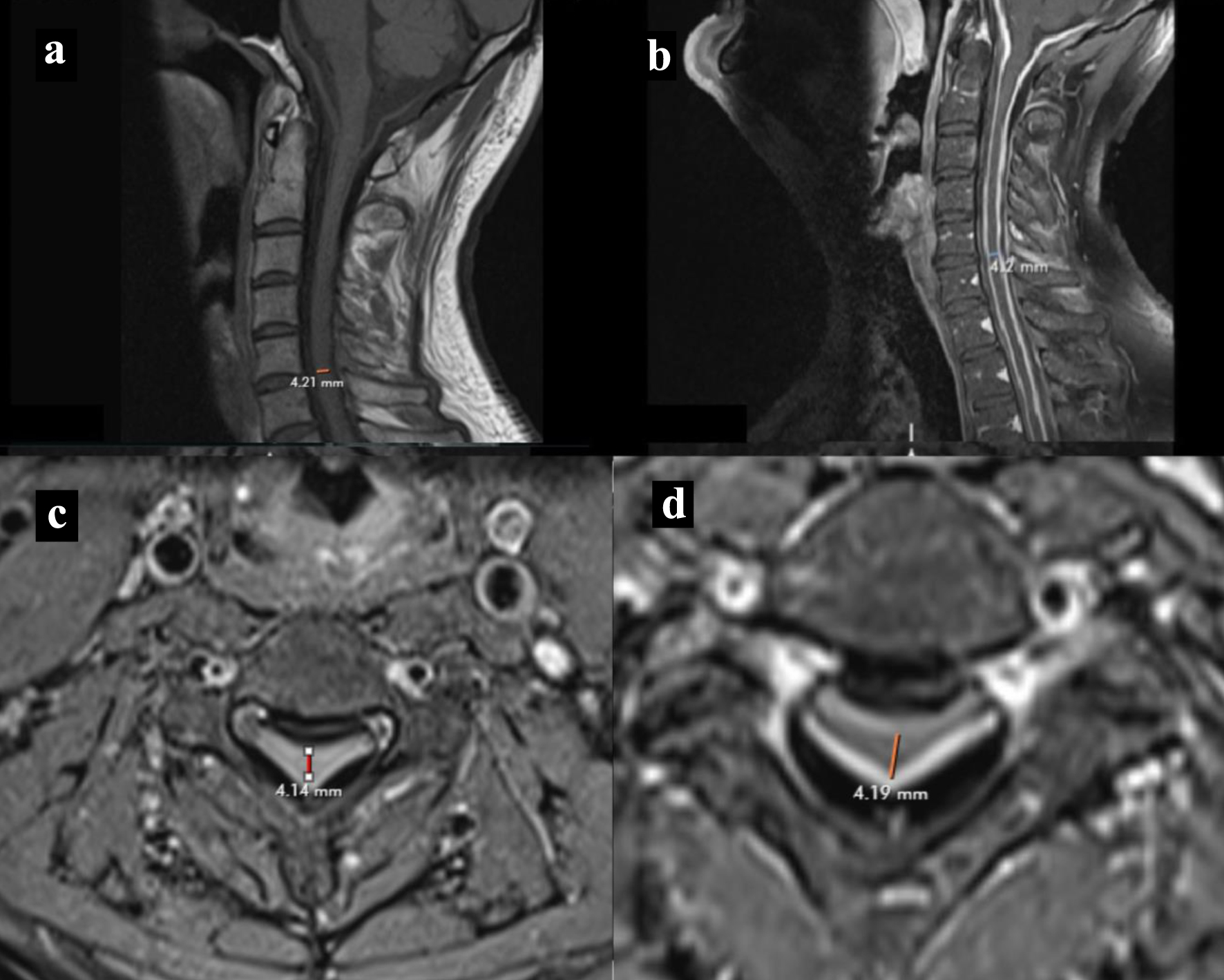 Click for large image | Figure 2. MRI cervical spine. (a, b) T1-weighted MRI spine sagittal section without and with contrast showing cervical spine dural thickening with narrowing of cord respectively. (c, d) T1-weighted MRI spine without and with contrast axial section showing narrowing of cord respectively. MRI: magnetic resonance imaging. |
Cerebrospinal fluid (CSF) showed lymphocytic pleocytosis with very high protein (Table 1). Patient’s Quantiferon gold test was positive. However, his infectious workup including acid-fast bacilli (AFB) stain and cultures were negative. He was later seen by our infectious disease colleagues and advised against anti-tuberculous therapy. Further workup for rheumatological and other autoimmune causes including erythrocytic sedimentation rate (ESR), C-reactive protein (CRP), antineutrophilic antibody (ANA), antineutrophil cytoplasmic antibody (ANCA), rheumatoid arthritis (RA) factor, anti-Sjogren syndrome A/B (anti-SSA/SSB), and angiotensin converting enzyme (ACE) levels were normal or negative except slightly elevated serum immunoglobulin-4 (IgG4) levels. CT of chest did not reveal any evidence of pulmonary fibrosis or nodules.
 Click to view | Table 1. CSF Analysis |
Diagnosis
Based on the imaging findings of diffuse dural thickening in the brain and spinal cord, extensively at cervical region, the presenting symptoms of cervical cord compression were highly likely. The patient’s history of ankylosing spondylitis made the diagnosis challenging, but his MRI showed no spondylitic changes at cervical levels, which emphasizes the need to consider other causes of his spinal cord compression in this case. Initially, tuberculous meningitis was patient’s presumed diagnosis for 9 years. Findings from CSF studies, like lymphocytic pleocytosis, elevated protein levels and positive Quantiferon gold test supported his diagnosis of tuberculous meningitis. However, further infectious workup including AFB staining and microbial TB and bacterial cultures were negative. Moreover, there were no diagnostic findings on CT chest to suggest the diagnosis of tuberculosis or sarcoidosis. Inflammatory markers were negative except slightly elevated serum IgG4 level (104.4). MRI of the brain and spine showing diffuse dural thickening in the posterior fossa and entire spine, strongly supported the diagnosis of HP. Finally, all of these evidences put together with responsiveness to steroid therapy supported his diagnosis of cervical cord compression due to HP, possibly idiopathic as all other causes have been excluded.
Treatment and follow-up
Our patient was administered oral steroid therapy, specifically 40 mg of prednisone due to inadequate response to low-dose treatment with 20 mg of prednisone. He exhibited clinical improvement of his symptoms, and serial MRI showed a decrease in meningeal enhancement. The dural thickening, while non-enhancing, was less prominent compared to previous MRIs (Fig. 3). Although additional immunosuppressive therapy was considered, it was not implemented due to his positive response to steroids. After 6 months of higher dose steroid treatment, the dose was gradually tapered to 20 mg of prednisone and eventually discontinued after 7 to 8 months, in light of his clinical improvement. The patient was evaluated by neurosurgery, and a biopsy was not recommended, possibly due to the observed clinical improvement. Although he did experience occasional fatigue, weakness, and heaviness in his left arm but no new symptoms were reported and he resumed playing tennis and exercising.
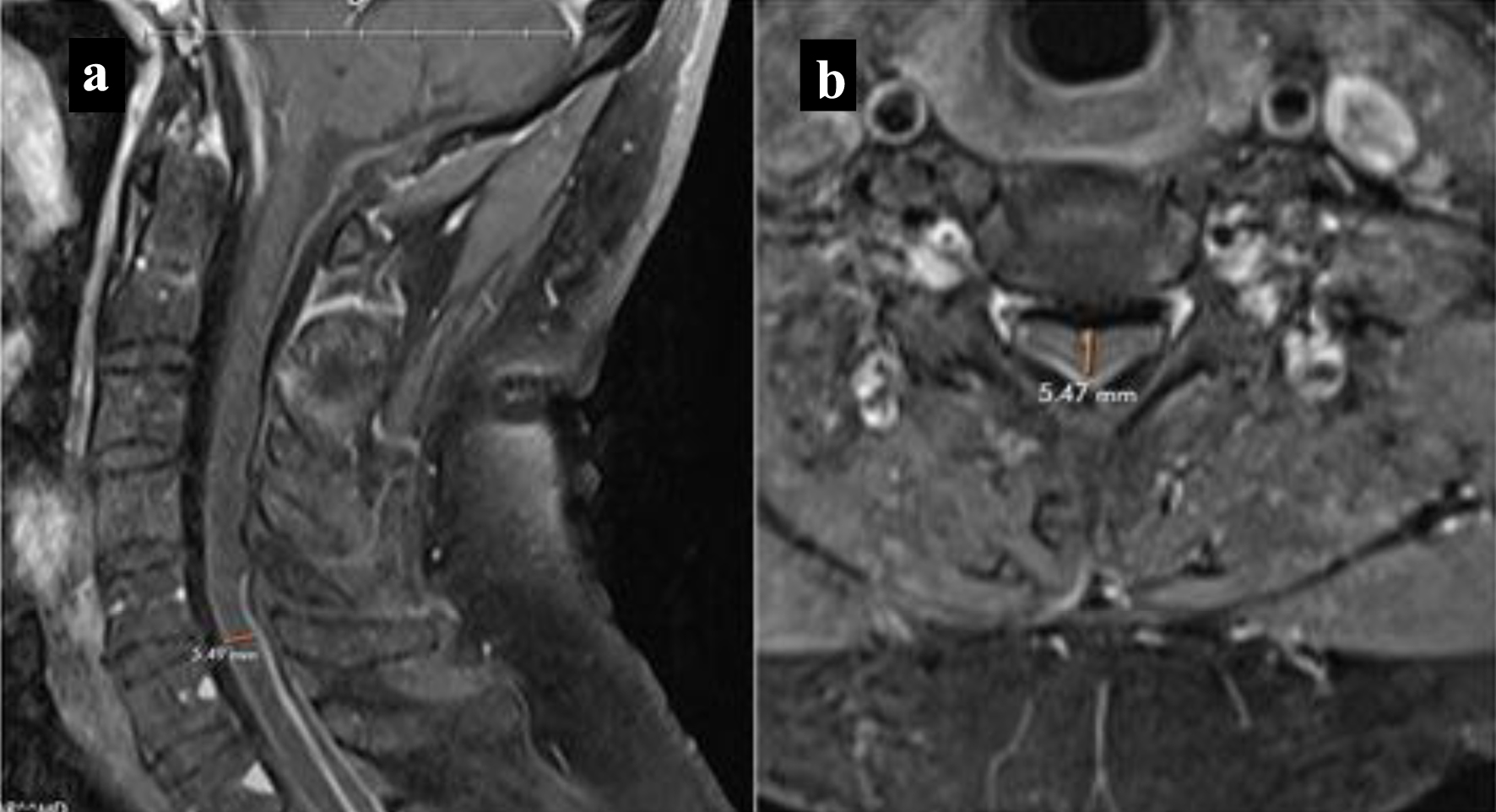 Click for large image | Figure 3. MRI cervical spine. Follow-up T1-weighted MRI with contrast showing decreased meningeal enhancement and narrowing. (a) Sagittal view. (b) Axial view. MRI: magnetic resonance imaging. |
Review of current literature
We did a literature search on PubMed for published cases of IHSP since 2009 and found a total of 22 cases. Findings from these studies have been described in Supplementary Material 1 (www.journalmc.org) [4-22].
We analyzed a total of 23 cases including ours. Mean age for presentation is 51 years ranging from 19 to 79 years with 65% (n = 15) female preponderance. Patients with IHSP usually have progressive presentation (83%) most commonly with weakness of limbs or extremities (78%) followed by sensory symptoms (61%), back or neck pain (52%), ataxia (30%) and bowel and bladder problems (26%). On examination, 48% (n = 11) patients revealed clinical features of myelopathy like hyperreflexia, clonus, Babinski sign and sensory level indicating spinal cord involvement.
Studies have reported elevated inflammatory markers in IHSP patients. Serum analysis showed elevated ESR in 35% (n = 8), CRP in 30% (n = 7) and RA in 9% (n = 2) patients. Furthermore, CSF analysis of four patients revealed elevated protein (> 45 mg/dL) along with elevated CSF leukocyte count with lymphocyte predominance in five patients. IHSP patients showed only cervical and thoracic spinal cord involvement with an average of 5 - 6 spinal segments. Nearly 36% (n = 8/22) patients reported circumferential dural thickening followed by posterior in 27% (n = 6), anterior in 23% (n = 5) and, both anterior and posterior in 14% (n = 3) patients. Nearly all patients showed hypointensity on T1- and T2-weighted imaging (except isointensity in two cases) with contrast enhancement. Biopsy of dural thickening revealed chronic non-specific inflammation with fibrosis, infiltrated with fibroblast, lymphocyte and plasma cells in all cases.
From 21 cases where treatment was reported, 71% (n = 15) patients had both surgical and conservative management either during initial or relapse management. Decompressive laminectomy is the preferred surgical approach while drugs like methylprednisolone, prednisone and dexamethasone were tried for conservative management. Fifty-two percent (n = 11) patients completely improved with the preferred treatment approach while only 19% (n = 4) showed no improvement. Six cases reported to have relapsing disease within an average 9-week period.
| Discussion | ▴Top |
We describe a case of IHSP with complete remission after steroids without any relapse. From the review of literature, only Fakuda et al have described a similar case of a 51-year-old male with IHSP who presented with radiculopathy and responded to steroids without any further relapses [6]. Apart from idiopathic causes, other possible etiologies for HP include autoimmune, infections and neoplasm (Table 2 [23-25]).
Click to view | Table 2. Causes of Hypertrophic Pachymeningitis |
Spinal form of HP is one of the rare causes of radiculomyelopathy. According to Charcot and Joffroy [26], hypertrophic spinal pachymeningitis (HSP) can be grouped into three stages on the basis of disease progression from radicular pain in neck or back followed by radiculopathy and lasting into myelopathy, attributed to vascular compromise and spinal nerve root or cord compression [26].
Demographic and clinical features
Mean age of presentation for IHSP is 51 years with higher female prevalence (M:F = 1:2). This is in concordance with the results reported by previous studies [11, 16]. IHSP most commonly presents as weakness, sensory loss, varying with neck or back pain, ataxia to bowel and bladder involvement, depending on the involvement of spinal nerve roots and cord compression. Qin et al [16] also reported weakness and sensory loss as most common presentations of IHSP in their review of biopsy proven cases. Ninety percent of the patients with clinical signs of myelopathy had progressive course of disease (Supplementary Material 1, www.journalmc.org). Our patient also progressed from sensory loss to weakness in bilateral arms, but no upper motor neuron (UMN) signs, only suggestive of radiculopathy ascribed to its nerve root compression. This shows that timely identification of IHSP presenting with common symptoms like weakness, sensory loss and even neck pain can prevent it from progression and lending into myelopathy. Elsberg’s [27] observations also highlight the importance of considering HSP as a potential diagnosis when clinical features suggestive of compression of three or more spinal nerve roots are present. Other spinal cord pathologies like extramedullary tumor, subdural hematoma, epidural abscess and neoplasm [11, 28, 29] can also imitate IHSP leading to its delay in diagnosis and timely intervention.
Laboratory features
During our comprehensive review, we identified total of four and five cases with elevated CSF proteins [16, 17, 21] and lymphocyte count [4, 16, 17, 21] respectively, including our case. Severity of CSF abnormalities including CSF protein and cell count is positively related to disease progression, as stated by Qin et al in their study [16]. Kupersmith et al have also noted more diffuse dural involvement in patients with abnormal CSF protein [23]. Similar to above studies, CSF findings in our patient also worsened as his disease progressed, further endorsing their positive correlation. Worsening of CSF protein elevation might be attributed to blockage of CSF circulation due to compression by duramater thickening. Further, increased vascular permeability in chronic inflammation can also lead to elevated protein and lymphocyte levels [16].
Among eight patients where signs of active inflammation like elevated ESR and CRP were observed and outcomes were reported, 63% (n = 5) patients either exhibited no clinical improvement or partially improved (Table 3) [7, 8, 11, 15, 20]. A study by Takahashi et al also documented no radiological improvement and delayed clinical development after a span of 2 years [19]. Consequently, patients with elevated inflammatory markers displayed worse prognosis in comparison to those without. In contrast, our patient did not show any signs of active inflammation and attained complete improvement with steroid therapy, aligning with the findings reported in the literature. Elevated inflammatory markers display continuous disease activity or active inflammation causing more duramater thickening and increased compression leading to disease severity. Similarly, persistent inflammation may hinder the effectiveness of disease treatment and resulting in bad prognosis [28].
Click to view | Table 3. Outcomes of IHSP With Different Clinical Variables |
Radiological and histopathology features
We only established cervical and thoracic cord involvement in IHSP from our review of cases with an average of 5 - 6 spinal segments. This is consistent with the findings reported previously in the literature [8, 11, 16, 18]. IHSP was most commonly found in circumferential (36%) followed by posterior and anterior location (Supplementary Material 1, www.journalmc.org). This is in contrast to reports from previous studies where they found higher percentages of circumferential involvement with minimal or no posterior cord involvement [11, 16]. These differences in between the findings of current study with previous highlight the variability of distribution in IHSP lesion within the spinal cord and have implication for further understanding the disease pathophysiology and clinical presentation.
MRI can be a useful diagnostic modality in establishing IHSP. It usually shows hypointense in T1- and T2-weighted imaging with gadolinium contrast enhancement. On T2-weighted imaging, low intensity central mass represents fibrous tissue of duramater and peripheral enhancement describes hypervascularity and inflammatory infiltrates [30, 31]. However, these MRI features are not peculiar in differentiating it from other space occupying lesions like epidural abscess and hematoma [15]. Histopathological findings in correlation with radiological features help in substantiating diagnosis of IHSP and ruling out other possible etiologies. It typically shows fibrous collagen tissues along with chronic non-specific inflammation infiltrated with predominantly lymphocyte and plasma cell. Immunohistochemistry also revealed CD3- and CD20-positive T and B lymphocytes, respectively [18, 21].
Treatment outlines and relapses
Laminectomy with cord decompression and steroids are mainstay treatment therapy for IHSP. From our review of literature, both surgery and steroids were used for initial management in 13 patients. Thirty-one percent of these patients did not show any improvement after the treatment (Table 3). Longer duration of clinical symptoms, absence of post-surgical continuous steroids therapy and preceding spinal cord injury have been reasonably explained for poor prognosis of disease in these cases [8, 16, 18]. Conservative management alone in the form of steroid therapy was tried in four cases and all of them completely improved; however, 50% of them showed recurrence of disease (Table 3) (Supplementary Material 1, www.journalmc.org). Presence of myelopathy features or UMN signs could explain the rationale behind their relapse [4, 17]. On the contrary, steroids alone can be an effective management for patients with symptoms of radiculopathy including current case. In certain studies, methotrexate as an alternative immunosuppressive agent has been employed, specifically during relapse of disease, yielding more favorable treatment outcomes [14, 20]. Surgery without steroids was used as initial management in four patients where one had partial improvement [5] and one patient relapsed after 12 weeks [20]. Surgical management without steroids can have commending results depending on individual patient’s characteristics, disease severity and stages; however, patients with long-standing disease path with significant fibrosis and edema often require a steroid course after surgical debulking as they help to attain complete remission and decrease persistent inflammation around cord [12, 19, 21].
Relapse of IHSP remains a critical aspect of disease management with poor understanding of its implications and underlying mechanism. From our review of cases, six patients experienced relapse with an average 9-week period [4, 8, 14, 16, 17, 20]. Some studies suggest patients with elevated inflammatory markers and leukocytosis have increased relapse rate in comparison to those without these findings [5, 16]. Elevated inflammatory markers manifest active dural inflammation that is present before surgical management [28]. Kim and Park have reported relapse of IHSP in their studies with elevated inflammatory markers; however, this does not hold true in all cases [8, 20]. There have been speculations about residual pachymeningitis and arachnoiditis associated with disease relapse in cases where only posterior duramater is excised or removed during surgery due to difficulty in reaching to anterior location. The remaining inflammatory process can extend in any direction to involve the internal duramater causing recurrence of pachymeningitis [28]. This review also reveals recurrence of IHSP only in cases where anterior or circumferential dural involvement was found, further consolidating this hypothesis [4, 14, 16, 17, 20]. Understanding the association between dural extent and the likelihood of relapse is crucial for refining treatment approaches and preventing recurrence of IHSP.
Learning points
IHSP is rare progressive inflammatory spinal disease causing radiculomyelopathy. It is important to consider IHSP as a differential with other space occupying lesion in spinal cord.
MRI along with histopathology can aid in establishing the confirmatory diagnosis, while elevated CSF markers can give valuable insight about disease progression.
Steroids alone can be considered sufficient for patients without clinical myelopathy features; however, surgery followed by steroid should be considered as a definitive line of management in all other cases of IHSP.
Patients with circumferential and anterior cord involvement and elevated inflammatory markers should be followed long term as they have higher chances for disease recurrence.
The overall outcomes of IHSP appear to be independent of any particular variable, encompassing a composite of various clinical, laboratory, radiological findings and treatment approaches.
| Supplementary Material | ▴Top |
Suppl 1. Literature search on PubMed for published cases of IHSP since 2009.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Informed consent has been obtained from the patient for submission of this case report.
Author Contributions
RH, RDS, SS and RKG treated the patient during her inpatients and outpatients visits. AG and DU analyzed and interpreted the clinical data and performed the review of the literature with close supervision by RKG. RS performed MRI of the brain and spine. All authors read and approved the final manuscript.
Data Availability
The data generated and analyzed during this study are available in Supplementary Material 1.
| References | ▴Top |
- Hassan KM, Deb P, Bhatoe HS. Idiopathic hypertrophic cranial pachymeningitis: Three biopsy-proven cases including one case with abdominal pseudotumor and review of the literature. Ann Indian Acad Neurol. 2011;14(3):189-193.
doi pubmed pmc - Slade SJ, Bauer EM, Stone VV, Dave AJ. Spinal IgG4-related hypertrophic pachymeningitis with spinal cord compression: case report and literature review. World Neurosurg. 2019;130:65-70.
doi pubmed - Hahn LD, Fulbright R, Baehring JM. Hypertrophic pachymeningitis. J Neurol Sci. 2016;367:278-283.
doi pubmed - Alsulaiman A. Idiopathic hypertrophic spinal pachymeningitis: a diagnostic challenge: a case report and review of the literature. J Neurosci Rural Pract. 2020;11(1):175-177.
doi pubmed pmc - Bang JH, Cho KT, Kim EJ. Idiopathic Hypertrophic Pachymeningitis in the Craniocervical Junction. Korean J Spine. 2015;12(3):169-172.
doi pubmed pmc - Fukuda A, Punaro E, Rogerio F, de Souza Queiroz L, Reis F. Idiopathic hypertrophic pachymeningitis as a rare cause of cervical compressive myelopathy. J Craniovertebr Junction Spine. 2017;8(4):387-389.
doi pubmed pmc - Jee TK, Lee SH, Kim ES, Eoh W. Idiopathic hypertrophic spinal pachymeningitis with an osteolytic lesion. J Korean Neurosurg Soc. 2014;56(2):162-165.
doi pubmed pmc - Kim JH, Park YM, Chin DK. Idiopathic hypertrophic spinal pachymeningitis: report of two cases and review of the literature. J Korean Neurosurg Soc. 2011;50(4):392-395.
doi pubmed pmc - Lowden MR, Gill D. Teaching NeuroImage: idiopathic hypertrophic spinal pachymeningitis. Neurology. 2009;72(5):e27.
doi pubmed - Araujo Melo LL, Daher MT, Goncalves MVM, Freitas MB. Idiopathic hypertrophic spinal pachymeningitis: a case report. Rev Bras Ortop (Sao Paulo). 2022;57(3):521-523.
doi pubmed pmc - Olubajo F, Yermakova T, Highley JR, Arzoglou V. Concomitant idiopathic hypertrophic spinal pachymeningitis and Guillain-Barre syndrome in a patient: coincidence or a triggering mechanism? J Neurosurg Spine. 2017;27(3):335-340.
doi pubmed - Chen H, Li Y, Mehra S, et al. Idiopathic hypertrophic pachymeningitis as a rare cause of spinal cord compression. Journal of Medical Cases, North America. 2012;4:267-269.
- Hsu HT, Hsu SS, Chien CC, Lai PH. Teaching NeuroImages: Idiopathic hypertrophic spinal pachymeningitis mimicking epidural lymphoma. Neurology. 2015;84(9):e67-68.
doi pubmed - van der Pol CB, Chakraborty S, Cote I, Humphrey-Murto S, Michaud J. Case 216: hypertrophic spinal pachymeningitis. Radiology. 2015;275(1):303-307.
doi pubmed - Park JY, Choi I, Khil EK, Kim WJ, Shin IY. Idiopathic hypertrophic spinal pachymeningitis with spinal cord lesion: a case report. Korean J Neurotrauma. 2020;16(2):367-373.
doi pubmed pmc - Qin LX, Wang CY, Hu ZP, Zeng LW, Tan LM, Zhang HN. Idiopathic hypertrophic spinal pachymeningitis: a case report and review of literature. Eur Spine J. 2015;24(Suppl 4):S636-643.
doi pubmed - Kutty RK, Sreemathyamma SB, Sivanandapanicker JL, Peethambaran A. Idiopathic hypertrophic spinal pachymeningitis: a rare cause of spinal cord compression. Neurol India. 2019;67(5):1380-1385.
doi pubmed - Ranasinghe MG, Zalatimo O, Rizk E, Specht CS, Reiter GT, Harbaugh RE, Sheehan J. Idiopathic hypertrophic spinal pachymeningitis. J Neurosurg Spine. 2011;15(2):195-201.
doi pubmed - Takahashi H, Wada A, Yokoyama Y, Ishii M, Shibuya K, Suguro T. Idiopathic hypertrophic spinal pachymeningitis: a case report. J Orthop Surg (Hong Kong). 2010;18(1):113-117.
doi pubmed - Park TJ, Seo WD, Kim SY, Cho JH, Kim DH, Kim KH. Effective response of methotrexate for recurrent idiopathic hypertrophic spinal pachymeningitis. Korean J Spine. 2016;13(4):200-203.
doi pubmed pmc - Tosa M, Hara M, Morita A, Ninomiya S, Ebashi M, Kamei S, Maseda M, et al. Idiopathic hypertrophic spinal pachymeningitis. Intern Med. 2015;54(15):1923-1926.
doi pubmed - Maheshwari V, Gill M, Kumar M, Srivastava C. Idiopathic hypertrophic cervical pachymeningitis - A rare report. Neurol India. 2016;64(5):1038-1040.
doi pubmed - Kupersmith MJ, Martin V, Heller G, Shah A, Mitnick HJ. Idiopathic hypertrophic pachymeningitis. Neurology. 2004;62(5):686-694.
doi pubmed - Abrantes FF, Moraes MPM, Rezende Filho FM, Pedroso JL, Barsottini OGP. A clinical approach to hypertrophic pachymeningitis. Arq Neuropsiquiatr. 2020;78(12):797-804.
doi pubmed - Xiao X, Fu D, Feng L. Hypertrophic pachymeningitis in a southern chinese population: a retrospective study. Front Neurol. 2020;11:565088.
doi pubmed pmc - Charcot JM, Joffroy A. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux anterolateraux de la moelle epiniere.
- Elsberg CA. Tumor of the spinal cord. New York: PB Hoeber; 1925. p. 342-343
- Ito Z, Osawa Y, Matsuyama Y, Aoki T, Harada A, Ishiguro N. Recurrence of hypertrophic spinal pachymeningitis. Report of two cases and review of the literature. J Neurosurg Spine. 2006;4(6):509-513.
doi pubmed - Ezzeldin M, Shawagfeh A, Schnadig V, Smith RG, Fang X. Hypertrophic spinal pachymeningitis: idiopathic vs. IgG4-related. J Neurol Sci. 2014;347(1-2):398-400.
doi pubmed - Mamelak AN, Kelly WM, Davis RL, Rosenblum ML. Idiopathic hypertrophic cranial pachymeningitis. Report of three cases. J Neurosurg. 1993;79(2):270-276.
doi pubmed - Nishizaki T, Iwamoto F, Uesugi S, Akimura T, Yamashita K, Ito H. Idiopathic cranial pachymeningoencephalitis focally affecting the parietal dura mater and adjacent brain parenchyma: case report. Neurosurgery. 1997;40(4):840-843; discussion 843.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.