

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 14, Number 2, February 2023, pages 64-70

Statin-Induced Immune-Mediated Necrotizing Myopathy Resulting in Proximal Muscle Weakness
Mohammed S. Abdallaa, c, Qishuo Zhanga, Monzer O. Abdallaa, Suhair S. Abdel-Jalilb
aDepartment of Internal Medicine, Ascension Saint Francis Hospital, Evanston, IL, USA
bDepartment of Rheumatology, Ascension Saint Francis Hospital, Evanston, IL, USA
cCorresponding Author: Mohammed S. Abdalla, Department of Internal Medicine, Ascension Saint Francis Hospital, Evanston, IL 60202, USA
Manuscript submitted December 14, 2022, accepted January 28, 2023, published online February 25, 2023
Short title: Statin-Induced IMNM
doi: https://doi.org/10.14740/jmc4039
| Abstract | ▴Top |
Statin-induced immune-mediated necrotizing myopathy (IMNM) is a subtype of IMNM linked to exposure to statins and is characterized by positive anti-hydroxymethylglutaryl (HMG) coenzyme A reductase (HMGCR) antibodies. Although rare, this entity has become increasingly recognized as a cause of proximal muscle weakness, especially with the widespread use of statin therapy. Unlike typical statin-associated muscle symptoms, IMNM myopathy often causes severe muscle injury, and muscle weakness persists or sometimes worsens following the withdrawal of statin therapy. Medical practitioners need to keep a high index of clinical suspicion for statin-induced IMNM in patients taking statins who present with muscle weakness. The disease can be debilitating, and treatment strategies are not well established despite the advances that have been made in the diagnosis. Here we present the clinical characteristics and disease course of two cases of statin-induced IMNM. Both patients presented with progressive proximal muscle weakness and myalgias while on long-term statin therapy without significant improvement in their symptoms following the withdrawal of statin therapy. IMNM was suspected, and both patients were found to have high titers of anti-HMG coenzyme A reductase antibodies and demonstrated microscopic features consistent with a diagnosis of IMNM on muscle biopsy. The patients experienced significant disability due to muscle weakness and required a protracted course of escalated immunosuppressive therapy. Although rare, IMNM should be suspected in patients taking statins who present with muscle weakness that fails to improve or worsens when statins were stopped. Early diagnosis and institution of immunosuppressive therapy are important to prevent the progression of the disease.
Keywords: Immune-mediated necrotizing myopathy; Statin-induced myopathy; Anti-HMG-CoA reductase antibodies
| Introduction | ▴Top |
Statin-associated muscle symptoms represent a broad spectrum of clinical manifestations ranging from nonspecific muscle aches to severe necrotizing myopathy. Immune-mediated necrotizing myopathy (IMNM) is an increasingly recognized muscle disease often linked to statin use and occurs in about two to three patients for every 100,000 individuals exposed to statins [1]. However, in certain populations, statin-induced IMNM is now the most commonly observed form (> 50% of all cases) of inflammatory muscle disease [2].
Unlike toxic myopathy commonly associated with statin use, statin-induced IMNM is associated with a much worse prognosis and requires timely diagnosis and institution of immunosuppressive therapy [3-5]. The pathogenesis of this entity is not fully understood. However, many studies have linked the disease to the induction of anti-hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase antibodies by statin use [6], and these antibodies are used as a diagnostic test [7]. In addition to positive anti-HMG-CoA reductase antibodies, the diagnosis of statin-induced IMNM is also made by demonstrating the unique microscopic features on muscle biopsy, namely, myofiber degeneration and necrosis out of proportion to the inflammatory infiltrate [8]. Treatment of this entity requires prolonged, often escalating courses of immunosuppressive therapy. In this communication, the authors present two cases of statin-induced IMNM, describing the clinical characteristics, diagnosis, and outcome of immunosuppressive therapy.
| Case Reports | ▴Top |
Case 1
Investigations
A 54-year-old female presented with progressive bilateral leg pain and weakness for 2 months. She reported difficulty climbing stairs, getting out of bed, or low seats without assistance.
Her past medical history includes diabetes mellitus, hyperlipidemia, and obesity. She takes metformin, glimepiride, and lisinopril. She has also been taking atorvastatin 20 mg daily for 2 years prior to her presentation. Her vital signs were within normal limits. Physical examination was notable for muscle tenderness and proximal muscle weakness, with muscle power of 3/5 of bilateral deltoids, hip flexors, and has weak neck flexors. Physical examination was otherwise non-contributory.
Diagnosis
Initial investigations include aspartate aminotransferase (AST): 91(13 - 39 IU/L), alanine aminotransferase ALT: 224 (7 - 52 IU/L), creatine phosphokinase (CPK): 6,523 IU/L (30.0 - 223.0 IU/L), aldolase: 49.5 U/L (1.2 - 7.6 U/L) erythrocyte sedimentation rate (ESR): 73 (0 - 30 mm/h) and thyroid-stimulating hormone (TSH): 2.85 (0.27 - 4.20 µIU/mL) and hemoglobin A1C: 6.4 (< 5.7%). Antinuclear antibodies (ANA) and rheumatoid factor (RF) were negative. Anti-HMG-CoA reductase antibodies immunoglobulin G (IgG) were elevated > 200 (0 - 19 U). Myositis extended panel was negative (Table 1). Electromyography (EMG) of the left leg was consistent with electrodiagnostic evidence of mild proximal myopathy.
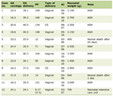 Click to view | Table 1. Myositis Extended Panel for Case 1 |
Muscle biopsy (Fig. 1) demonstrated the presence of scattered regenerating and necrotic muscle fibers without significant chronic lymphoid inflammation, consistent with a diagnosis of IMNM. In the context of statin use and positive HMG-CoA reductase antibodies, it was concluded that atorvastatin was the likely culprit.
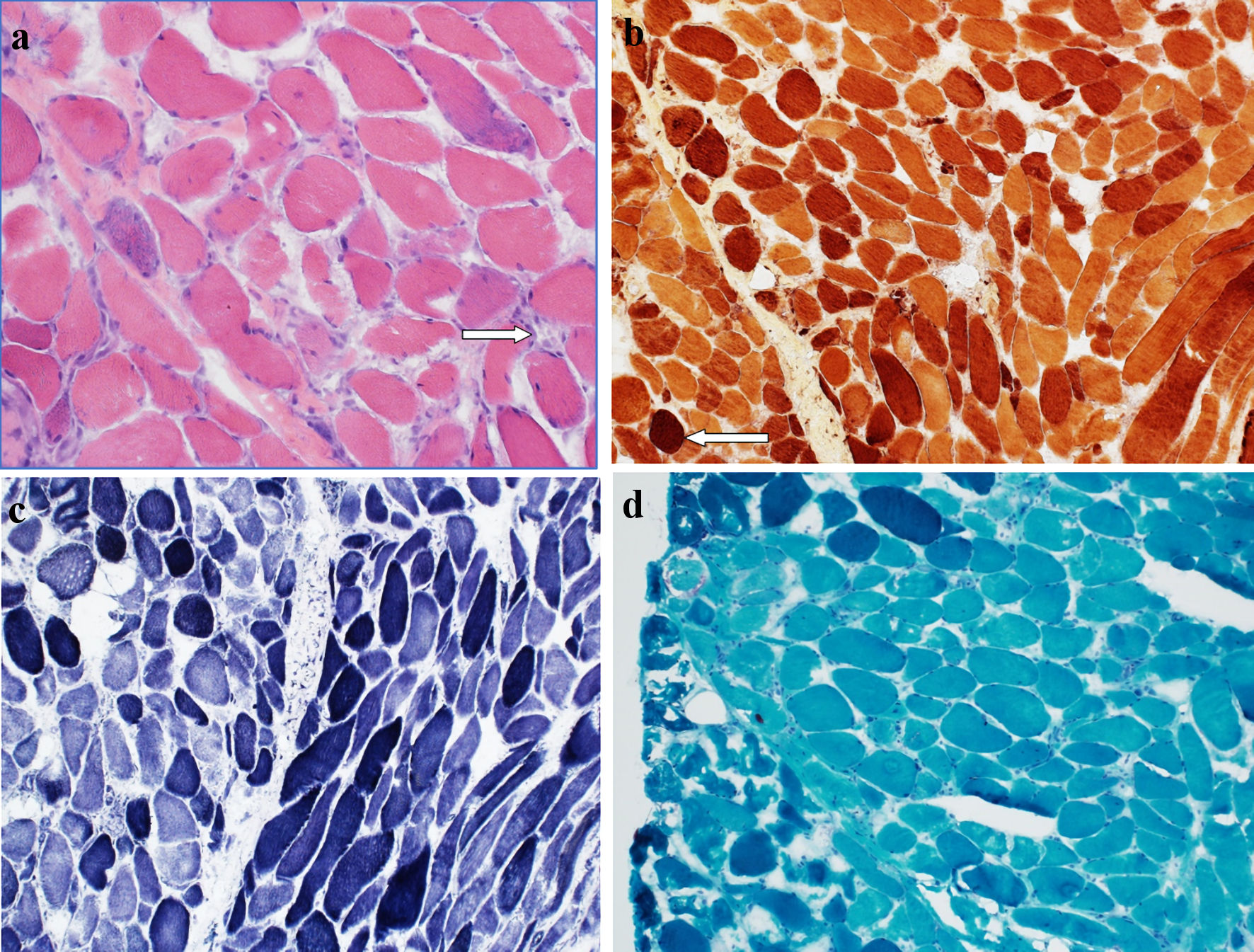 Click for large image | Figure 1. (a) Hematoxylin and eosin-stained section of muscle biopsy showing occasional regenerating and necrotic muscle fibers, with an associated sparse chronic lymphoid inflammation (arrow). No morphologic evidence of vasculitis is seen. No perifascicular atrophy is noted. (b) Esterase preparation darkly highlights macrophages (arrow) seen in association with necrotic myofibers. (c) NADH-TR preparation shows variable loss of the normal intermyofibrillar staining pattern in regenerating and necrotic muscle fibers. (d) No abnormal inclusions or rimmed vacuole type structures are noted on modified Gomori trichrome preparation. NADH-TR: nicotinamide adenine dinucleotide hydrogen-TR stain. |
Treatment
Statin therapy was stopped. The patient was started on oral prednisone 60 mg daily, and methotrexate 10 mg weekly.
Follow-up and outcomes
The patient experienced an improvement in her muscle pain after stopping atorvastatin, but her muscle weakness continues to worsen. However, she experienced a notable improvement in her symptoms a few weeks after the start of her immunosuppressive treatment with prednisone and methotrexate with no significant side effects at her first follow-up visit. She is scheduled for regular follow-up with her rheumatologist.
Case 2
Investigations
A 59-year-old male presented with 3 weeks of progressive muscle weakness involving his arms associated with difficulty lifting things above his head and reported some muscle stiffness that improves with physical activity. A few weeks later, the weakness extended to the lower extremities, with difficulty standing up from a sitting position. His medical history was significant for hyperlipidemia, type-2 diabetes mellitus, hypertension, and obesity. His long-term medications include insulin, lisinopril, metformin, and atorvastatin 80 mg daily. The patient was initially taking simvastatin for 6 years for hyperlipidemia, then was switched to atorvastatin 80 mg daily 5 years prior to this presentation. On physical examination, he had a symmetrical proximal muscle weakness with muscle power of 4/5, in both upper and lower extremities. His deep tendon reflexes were normal and symmetrical, and he did not have tremors or pronator drift. Physical examination was otherwise non-contributory.
Diagnosis
He was initially diagnosed with rhabdomyolysis considering a high creatine kinase (CK) of 18,315 (30.0 - 223.0 IU/L). His atorvastatin was held and treated with intravenous fluids with a gradual decrease in his CK level, which plateaued at 6,000. His initial labs were remarkable for AST: 378 (13 - 39 IU/L), ALT: 531 (7 - 52 IU/L), aldolase: 157 U/L (1.2 - 7.6 U/L), ESR: 12 (0 - 30 mm/h) and TSH: 1.897 (0.27 - 4.20 µIU/mL), lactate dehydrogenase 1,415 (140 - 271 U/L), and hemoglobin A1C 6.9% (< 5.7%). His urine was positive for blood without red blood cells (RBCs). Autoimmune screens, including ANA, RF, JO1 antibody, extractable nuclear antigen (ENA) SSA (RO) and ENA SSB (LA) antibodies, scleroderma SCL-70, and anti-neutrophil cytoplasmic antibodies IgG were all negative. Anti-hydroxymethylglutaryl (HMG) coenzyme A reductase (HMGCR) IgG antibody was elevated to more than 200 (0 - 19 U). Myositis extended panel was reported as negative (Table 2). The patient had EMG examination, which showed primary muscle disease without large fiber neuropathy. A muscle biopsy (Fig. 2) demonstrated necrotic muscle fibers with only sparse focal lymphoid inflammation consistent with a diagnosis of IMNM, likely induced by statin treatment.
Click to view | Table 2. Myositis Extended Panel for Case 2 |
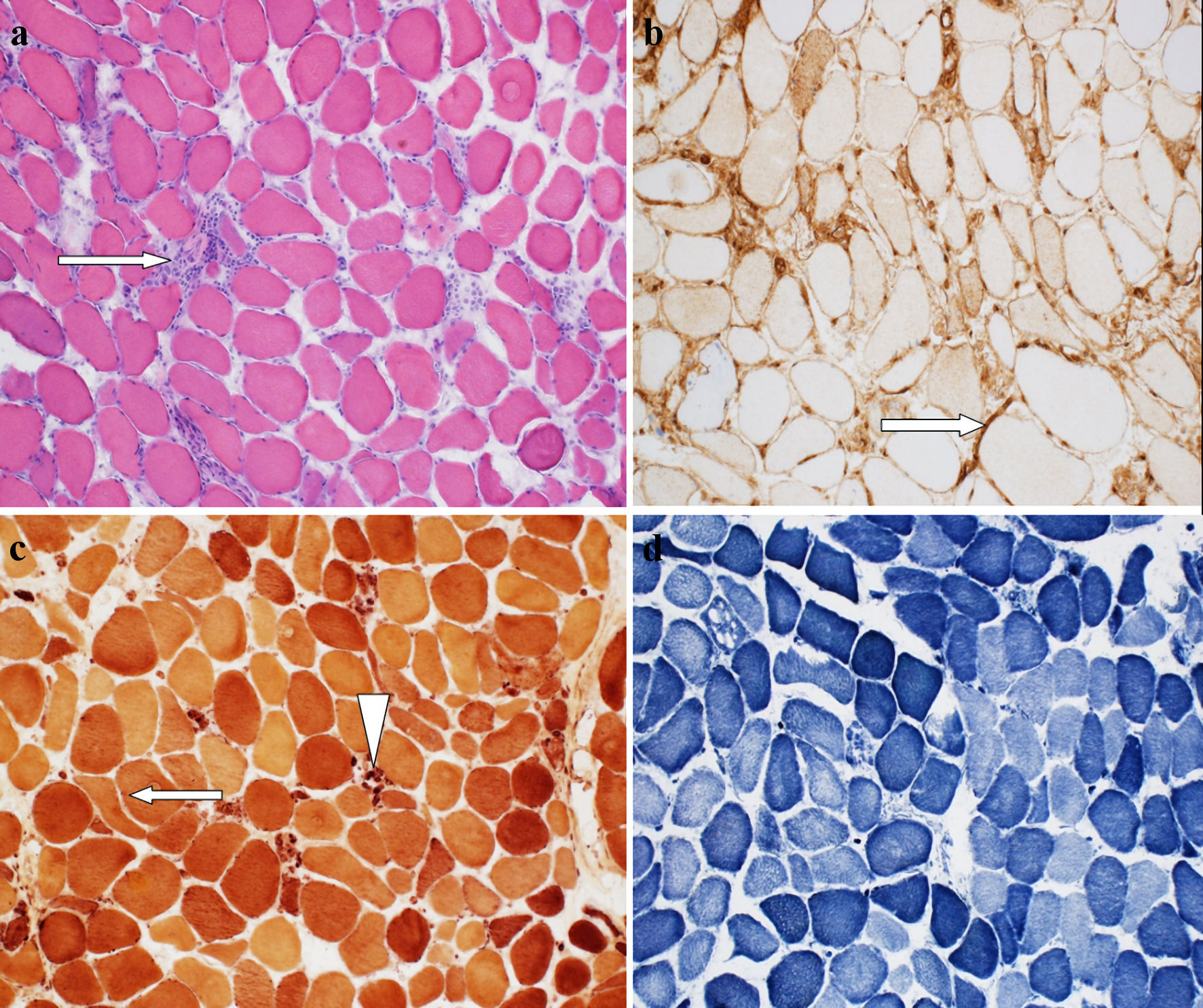 Click for large image | Figure 2. (a) Hematoxylin and eosin (H&E)-stained section of muscle biopsy demonstrates the presence of scattered regenerating and necrotic muscle fibers with only very sparse focal chronic lymphoid inflammation (arrow) (“necrotizing myopathy”). (b) MHC1 staining showing patchy mild to moderate increase in membranous muscle fiber staining for MHC1 (arrow). (c) Esterase preparation darkly highlights scattered acutely angulated atrophic muscle fibers (arrow), and highlights macrophages seen in association with necrotic muscle fibers (arrowhead). (d) NADH-TR preparation demonstrates variable loss of the normal intermyofibrillar staining pattern in regenerating and necrotic muscle fibers. NADH-TR: nicotinamide adenine dinucleotide hydrogen-TR stain; MHC: major histocompatibility complex. |
Treatment
Statin treatment was stopped, and the patient was started on oral prednisone and methotrexate.
Follow-up and outcomes
On follow-up 1 month later, he reported some improvement in muscle strength. He however had persistent difficulty standing up from a sitting position. He was then treated with three doses of pulsed intravenous methylprednisolone 1,000 mg with some improvement in muscle strength but still was unable to get out of bed unassisted. He was started on intravenous immunoglobulin (IVIG), with significant improvement in muscle strength. He then resumed oral prednisone with a reduced dose along with weekly methotrexate and remained on IVIG. His muscle weakness resolved, and he was back to baseline; he was able to return to work and remained almost fully functional 2 years after diagnosis.
| Discussion | ▴Top |
The increasing use of statins as first-line lipid-lowering agents has led to increasing reports of statin-associated muscle symptoms and unmasking a peculiar form of myopathy with an autoimmune nature associated with their use. Unlike patients with the commonly described statin-induced toxic myopathy, who typically improve after discontinuation of the offending medication, most patients with IMNM develop persistent muscle weakness and elevation of markers of muscle injury, namely CK, long after discontinuation of statins. Most of these patients require immunosuppressive therapies to show some clinical improvement. Thus, this distinct clinical entity, known as statin-associated autoimmune necrotizing myopathy, has become increasingly recognized [3-5].
The pathogenesis of statin-induced IMNM is not fully understood. The condition, typically positive for anti-HMGCR antibodies, is strongly associated with class II major histocompatibility complex (MHC) allele DRB1*11:01 and exposure to statins, suggesting a role for statin exposure in upregulating HMGCR in genetically susceptible patients [6]. Expression of HMGCR is upregulated not only in muscle tissue exposed to statins but also in regenerating muscle cells compared to resting myocytes which is felt to initiate an autoimmune response to muscle tissue [1]. The immune response is perpetuated through persistently increased HMGCR expression in regenerating muscle fibers long after cessation of statin therapy [9]. This autoimmune etiopathogenesis is further supported by the finding of MHC class I upregulation on the surface of non-necrotic muscle fibers in many of the reported cases [10]. In one case series, there was evidence of MHC class I antigen upregulation in 87.5% of the anti-HMGCR cases associated with the deposition of membrane attack complex (MAC), supporting a potentially complement-mediated immunopathogenesis [8]. Both of our patients tested positive for HMGCR antibodies. Interestingly, a recent study suggested that among different statins, atorvastatin was most strongly associated with the development of anti-HMG-CoA reductase myopathy when compared to simvastatin or rosuvastatin [11]. Both of our patients have been on long-term therapy with atorvastatin.
Anti-HMGCR IMNM presents clinically as a subacute or chronic progressive symmetrical proximal muscle weakness. Myalgia and dysphagia may occur in approximately one-third of patients [5, 12, 13]. The condition may not be initially clinically distinguishable from common statin-induced muscle symptoms at presentation. However, unlike toxic myopathy commonly associated with statin use, IMNM muscle weakness and myalgia do not typically improve and may even worsen after the withdrawal of statin therapy. Typical statin-induced muscle symptoms significantly improve or even resolve within weeks of cessation of statin therapy [5]. In addition, statin-induced necrotizing myopathy is often associated with significantly increased levels of CK, which typically persists after the withdrawal of statins, as seen in both of our cases.
Statin-induced necrotizing myopathy is associated with significantly increased levels of CK, a nonspecific marker of muscle injury. CK levels may also be linked to muscle strength and can be used as a surrogate marker for disease activity [13, 14]. Anti-HMGCR autoantibodies directed at the pharmacologic target of statins are a highly specific biomarker for the diagnosis of statin-induced IMNM [15]. In some studies, detecting these autoantibodies has shown sensitivity and specificity for the disease as high as 94.4% and 99.3%, respectively [7, 16, 17]. Many studies demonstrated a clear association between statin exposure and positive anti-HMGCR antibodies; one study showed that 67% of patients with positive HMGCR antibodies had a prior prescription for statins [10], and higher rates of statin exposure have been reported by other investigators [18]. Both of our patients had high titers of HMGCR antibodies.
Histologically, muscle biopsy in statin-induced IMNM is characterized by myofiber degeneration and necrosis out of proportion to the minimal lymphocytic infiltration, suggesting a lesser role of lymphocytes in the pathogenesis [8]. The inflammatory infiltrate consists primarily of macrophages in almost 100% of anti-HMGCR-associated myopathy biopsies. Most of these macrophages were typically positive for CD163 M2, indicating regeneration/repair, whereas a few cases (18%) had CD11c M1-positive macrophages characterized by phagocytic activity [8]. In contrast to dermatomyositis, where macrophages and plasmacytoid dendritic cells were typically located in the perimysium and perifascicular areas, these cells were characteristically distributed throughout the endomysium in HMGCR-associated myopathy [8].
There is no good literature about the use of immunosuppressants in IMNM as the disease entity is relatively new, described for the first time in 2004. Corticosteroids, oral prednisone, or pulsed intravenous methylprednisolone are the first-line therapy. Despite being a significant component of treatment, glucocorticoids are insufficient when used alone [19, 20]. Some experts recommend intravenous immunoglobulin as a first line, especially in pregnant patients and young patients [21]. According to the 224th ENMC International Workshop recommendations [22], initial treatment for IMNM should include intravenous or oral steroids, along with the addition of another agent at the same time or within 1 month, depending on disease severity and the initial response to treatment. Methotrexate is considered a reasonable option. IVIG could be added instead of, or in severe cases, along with methotrexate. IVIG should always be added within 6 months if other strategies are ineffective. Some experts recommended that IVIG be considered monotherapy in patients who cannot take high-dose steroids. In critically ill patients and patients with refractory disease, some experts recommended using plasma exchange, cyclophosphamide, and/or cyclosporine. Rituximab is increasingly used for severe or recalcitrant statin-induced autoimmune necrotizing myopathy, usually employing the rheumatoid arthritis protocol, and appears to be very effective [5, 6, 23].
After achieving maximum benefit from treatment, steroids should be tapered as tolerated. Only after at least 2 years of well-controlled disease with minimal or no steroids should other agents be stopped or tapered as tolerated. Many patients, however, may require chronic IVIG.
In some studies, early IVIG was associated with clinical improvement in the short-term follow-up in IMNM as measured by gains in muscle strength during follow-up. The clinical effect was especially significant in those with moderate to severe muscle weakness. Some factors, including female gender, older age, and initial oral or intravenous steroids, were associated with better biochemical improvement [24].
Learning points
Statin-induced IMNM is an uncommon form of myopathy that requires a high index of clinical suspicion, especially in patients treated with statins who develop muscle symptoms and fail to improve or progress after cessation of statin therapy. Although the condition can mimic other inflammatory myopathies, it has different pathophysiologic, morphologic, diagnostic, and therapeutic characteristics. Early recognition is vital to prevent progressive muscle damage and disability, and treatment requires immunosuppressive therapy. Despite treatment, many patients develop clinical relapses upon weaning from immunosuppressive therapy. Moreover, many patients require escalation of immunosuppressive therapy to different classes of drugs with the associated risks of immunosuppression. Further research into therapeutic options is required.
Acknowledgments
The authors would like to thank the Department of Pathology at Ascension Saint Francis Hospital for their input and support.
Financial Disclosure
No funding was received for this case report.
Conflict of Interest
There is no conflict of interest in this case report from all authors.
Informed Consent
Both patients provided consent for publication.
Author Contributions
MSA prepared the discussion part, reviewed the literature, provided relevant referencing, and obtained copy of the biopsy slides. QZ prepared the first case presentation including the patient’s history, physical examination, relevant investigations, treatment, and outcomes. MOA prepared the second case presentation. SAJ is the supervising rheumatologist who diagnosed the cases and helped with identifying the relevant clinical data and revision of the overall case report.
Data Availability
Any inquiries regarding supporting data availability of this the study should be directed to the corresponding author.
| References | ▴Top |
- Mammen AL. Statin-associated autoimmune myopathy. N Engl J Med. 2016;374(7):664-669.
doi pubmed - Muruganandam M, Iqbal A, Akpan EB, Dolomisiewicz AC, Waters YM, Emil NS, Nunez SE, et al. Statin-associated immune-mediated necrotizing myositis in Native Americans. Rheumatology (Oxford). 2022;61(12):4855-4862.
doi pubmed - Fauchais AL, Iba Ba J, Maurage P, Kyndt X, Bataille D, Hachulla E, Parent D, et al. [Polymyositis induced or associated with lipid-lowering drugs: five cases]. Rev Med Interne. 2004;25(4):294-298.
doi pubmed - Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord. 2007;17(2):194-200.
doi pubmed - Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve. 2010;41(2):185-190.
doi pubmed - Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Immune-Mediated Necrotizing Myopathy. Curr Rheumatol Rep. 2018;20(4):21.
doi pubmed - Mammen AL, Pak K, Williams EK, Brisson D, Coresh J, Selvin E, Gaudet D. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken). 2012;64(2):269-272.
doi pubmed - Chung T, Christopher-Stine L, Paik JJ, Corse A, Mammen AL. The composition of cellular infiltrates in anti-HMG-CoA reductase-associated myopathy. Muscle Nerve. 2015;52(2):189-195.
doi pubmed - Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, Casciola-Rosen LA. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63(3):713-721.
doi pubmed - Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62(9):2757-2766.
doi pubmed - Basharat P, Lahouti AH, Paik JJ, Albayda J, Pinal-Fernandez I, Bichile T, Lloyd TE, et al. Statin-Induced Anti-HMGCR-Associated Myopathy. J Am Coll Cardiol. 2016;68(2):234-235.
doi pubmed - Ramanathan S, Langguth D, Hardy TA, Garg N, Bundell C, Rojana-Udomsart A, Dale RC, et al. Clinical course and treatment of anti-HMGCR antibody-associated necrotizing autoimmune myopathy. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e96.
doi pubmed - Allenbach Y, Drouot L, Rigolet A, Charuel JL, Jouen F, Romero NB, Maisonobe T, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine (Baltimore). 2014;93(3):150-157.
doi pubmed - Werner JL, Christopher-Stine L, Ghazarian SR, Pak KS, Kus JE, Daya NR, Lloyd TE, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. 2012;64(12):4087-4093.
doi pubmed - Musset L, Allenbach Y, Benveniste O, Boyer O, Bossuyt X, Bentow C, Phillips J, et al. Anti-HMGCR antibodies as a biomarker for immune-mediated necrotizing myopathies: A history of statins and experience from a large international multi-center study. Autoimmun Rev. 2016;15(10):983-993.
doi pubmed - Mammen AL, Casciola-Rosen L, Christopher-Stine L, Lloyd TE, Wagner KR. Myositis-specific autoantibodies are specific for myositis compared to genetic muscle disease. Neurol Neuroimmunol Neuroinflamm. 2015;2(6):e172.
doi pubmed - Floyd JS, Brody JA, Tiniakou E, Psaty BM, Mammen A. Absence of anti-HMG-CoA reductase autoantibodies in severe self-limited statin-related myopathy. Muscle Nerve. 2016;54(1):142-144.
doi pubmed - Limaye V, Bundell C, Hollingsworth P, Rojana-Udomsart A, Mastaglia F, Blumbergs P, Lester S. Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle Nerve. 2015;52(2):196-203.
doi pubmed - Kassardjian CD, Lennon VA, Alfugham NB, Mahler M, Milone M. Clinical Features and Treatment Outcomes of Necrotizing Autoimmune Myopathy. JAMA Neurol. 2015;72(9):996-1003.
doi pubmed - de Souza JM, Hoff LS, Shinjo SK. Intravenous human immunoglobulin and/or methylprednisolone pulse therapies as a possible treat-to-target strategy in immune-mediated necrotizing myopathies. Rheumatol Int. 2019;39(7):1201-1212.
doi pubmed - Mammen AL, Tiniakou E. Intravenous Immune Globulin for Statin-Triggered Autoimmune Myopathy. N Engl J Med. 2015;373(17):1680-1682.
doi pubmed - Allenbach Y, Mammen AL, Benveniste O, Stenzel W, Immune-Mediated Necrotizing Myopathies Working G. 224th ENMC International Workshop: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14-16 October 2016. Neuromuscul Disord. 2018;28(1):87-99.
doi pubmed - Zhang W, Prince HM, Reardon K. Statin-induced anti-HMGCR antibody-related immune-mediated necrotising myositis achieving complete remission with rituximab. BMJ Case Rep. 2019;12(11):e232406.
doi pubmed - Wang JX, Wilkinson M, Oldmeadow C, Limaye V, Major G. Outcome predictors of immune-mediated necrotizing myopathy-a retrospective, multicentre study. Rheumatology (Oxford). 2022;61(9):3824-3829.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.