

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 13, Number 9, September 2022, pages 427-431

Immunocompetent Patient With Primary Bone Marrow Hodgkin Lymphoma
Armaan Dhaliwala, Vanessa F. Ellerb, Jeffrey J. Pua, b, c ![]()
aDepartment of Medicine, University of Arizona College of Medicine, Tucson, AZ, USA
bUniversity of Arizona NCI Designated Comprehensive Cancer Center, Tucson, AZ, USA
cCorresponding Author: Jeffrey J. Pu, Department of Medicine, University of Arizona College of Medicine, Tucson, AZ, USA
Manuscript submitted June 14, 2022, accepted August 4, 2022, published online September 28, 2022
Short title: Primary Bone Marrow Hodgkin’s Lymphoma
doi: https://doi.org/10.14740/jmc3973
| Abstract | ▴Top |
Hodgkin lymphoma (HL) is a hematologic malignancy that comprises about 10% of all lymphomas with the most common type being classical HL (cHL). The typical clinical presentation of cHL involves multiple region lymphadenopathy and a chest mass found on imaging. However, not all patients present with the typical symptomology of cHL which poses a diagnostic challenge. Extranodal HL, especially primary bone marrow HL (PBMHL), has been described in immunocompromised patients with human immunodeficiency virus (HIV). In this case report, we present a PBMHL case in an immunocompetent patient with no HIV exposure. We discuss a 51-year-old immunocompetent female who presented with 2 - 3 months of fever, confusion, generalized myalgias, and fatigue. She had no lymphadenopathy on physical exam. On further testing, the patient’s blood work demonstrated cytopenia and imaging confirmed no lymphadenopathy. Eventually, a bone marrow evaluation established her diagnosis of PBMHL. The patient expired after receiving one cycle of a modified chemotherapy regimen. This case illustrates that HL can be associated with an atypical clinical presentation which may delay diagnosis and treatment. PBMHL can occur in the normal population who is not immunocompromised nor HIV positive. In this situation, the best diagnostic approach is a thorough medical history, physical exam, and bone marrow aspiration and biopsy. Presence of constitutional symptoms without any lymphadenopathy or chest mass should raise the concern for possible atypical HL such as PBMHL. Accurate and timely identification of PBMHL allows for timely initiation of appropriate therapy. While cHL is responsive to chemotherapy, further research is required to improve the therapy for PBMHL.
Keywords: Hodgkin lymphoma; Atypical Hodgkin lymphoma; Primary bone marrow Hodgkin lymphoma; HIV
| Introduction | ▴Top |
Hodgkin lymphoma (HL) is a group of lymphoid malignancies characterized by the presence of Reed-Sternberg (RS) cells intermixed with non-neoplastic inflammatory cells. Thomas Hodgkin first described HL in an autopsy report in the 19th century. These lymphomas are divided into classical HL (cHL) and nodular lymphocyte-predominant HL based on morphological and immunophenotypic characteristics. cHL comprises about 90% of the HL cases and is further subdivided into four subtypes based on the pathologic features: nodular sclerosis cHL, mixed cellularity cHL, lymphocyte rich cHL, and lymphocyte depleted cHL.
RS cells are characterized with a rounded bilobed nuclei with a prominent eosinophilic nucleolus surrounded by a perinuclear halo and a weakly basophilic cytoplasm. RS cell surface strongly express CD30 and CD15 antigens but weakly express PAX5/BSAP antigens. RS cells also express PD-L1 and PD-L2, the programmed death ligands.
The typical clinical presentation of cHL involves asymptomatic lymphadenopathy or a mediastinal mass on a chest radiograph [1]. The lymph nodes of the neck comprising the cervical and/or supraclavicular nodes are the most frequently involved region of the disease [2]. Constitutional symptoms including fever, weight loss, and night sweats are commonly observed. Extranodal spread of HL, especially to the lungs, liver, bone, or bone marrow, can lead to a worse prognosis. However, some patients, especially those individuals with a history of human immunodeficiency virus (HIV) infection and immunosuppressed individuals, initially present atypical symptoms. Under such situations, patients could present with alcohol-associated pain, liver function test abnormalities, skin lesions, bone marrow infiltration causing unexplained cytopenia or bone pain, or paraneoplastic syndromes affecting the nervous system or leading to nephrotic syndrome. Usually, bone marrow involvement occurs in the advanced stages of the disease [3].
Primary bone marrow HL (PBMHL) is uncommon and most often described in HIV-positive patients. Isolated bone marrow HL present an aggressive clinical course and, unlike classical HL, display a poor response to the conventional HL treatments [3]. The current standard regimen for HL management in United States is ABVD (doxorubicin (adriamycin), bleomycin, vinblastine, and dacarbazine) combination chemotherapy. ABVD has been proven to be highly effective as initial therapy with less long-term toxicities when compared to alternate therapies [4]. An alternative chemotherapy regimen called escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisolone) has also been used for initial HL treatment or ABVD refractory HL treatment [5].
Here we present a unique PBMHL case, in which the patient presented with unexplained cytopenia and constitutional symptoms, without any sign of lymphadenopathy nor HIV positivity. The patient was diagnosed of HL via a bone marrow aspiration and biopsy.
All researches conducted in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
| Case Report | ▴Top |
Investigations
A 51-year-old HIV-negative female presented to the hospital with a 2-day history of fever, confusion, and a 3-month history of generalized myalgias and fatigue. She was originally from Somalia with a past medical history of polymyalgia rheumatica on intermittent prednisone treatment and chronic hyponatremia. Her blood work showed hemoglobin of 8.9 g/dL, white blood cell (WBC) count of 5.2 × 103/µL, and a platelet count of 60 × 103/µL. Her prothrombin time (PT)/international normalized ratio (INR) were mildly elevated at 20 s/1.7, serum sodium level was at 117 mmol/L. Her serum total bilirubin was 1.4 mg/dL, aspartate aminotransferase (AST) was 223 U/L, alanine aminotransferase (ALT) was 103 U/L, and alkaline phosphatase (ALP) was 198 U/L. A peripheral blood smear did not show any schistocytes, spherocytes, nor poikilocytes. The neutrophils showed toxic changes with pseudo-Pelger-Huet nuclei and vacuolization in the cytoplasm. Reticulocyte count was 6.6%, above the normal value range. Her hepatitis B and C panels were negative for either acute or chronic infection.
Her abdomen/pelvis computed tomography (CT) scan showed right-sided colitis and multiple hepatic lesions that represented either liver metastatic disease or microabscesses (Fig. 1). A magnetic resonance imaging (MRI) was conducted, but unable to distinguish these lesions. However, it revealed heterogenous enhancing bone marrow within the lumbar spine and pelvis. Her CT head was concerning for lucent lesions in the skull, and her MRI brain was suggestive of central pontine myelinolysis. Her disseminated intravascular coagulation (DIC) panel, ADAMTS13 activity, and autoimmune workup were all negative. Due to her altered mental status, a lumbar puncture was performed that yielded normal results except a cerebrospinal fluid (CSF) glucose of 96 mg/dL.
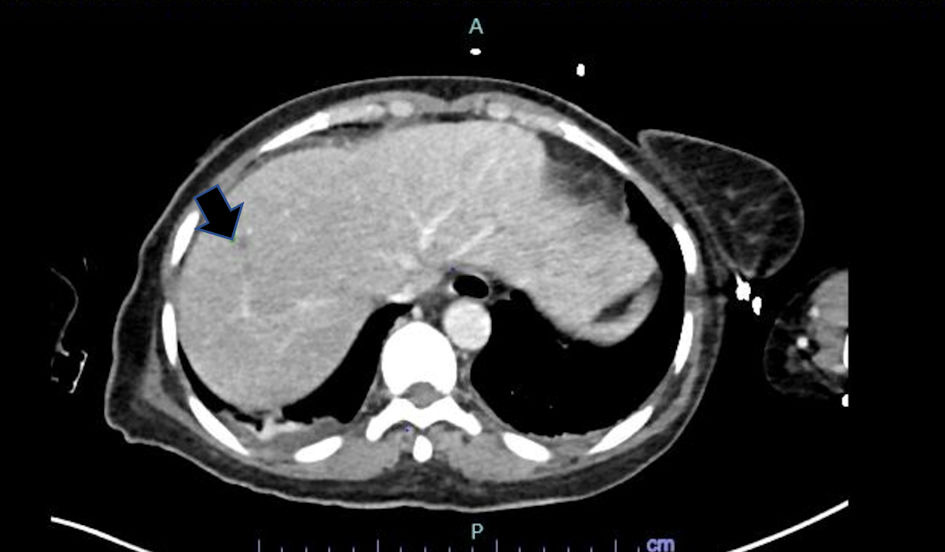 Click for large image | Figure 1. CT abdomen/pelvis demonstrated a small hypoattenuating lesion in the liver, measuring up to 7 mm. CT: computed tomography. |
Her iron panel was suggestive of anemia from chronic disease with a significantly elevation of ferritin (13,108 ng/mL). Her serum folate and vitamin B12 levels were normal. Her lactate dehydrogenase (LDH) level was elevated (547 U/L) and her haptoglobin level was reduced (< 10 mg/dL). Her inflammatory markers were significantly elevated with C-reactive protein (CRP) of 38 mg/L, erythrocyte sedimentation rate (ESR) of 60 mm/h, interleukin (IL)-2 of 84,250 pg/mL and chemokine (CXC motif) ligand 9 (CXCL9) of 11,741 pg/mL. Her Epstein-Barr virus (EBV) polymerase chain reaction (PCR) result was 1,400 IU/mL.
Diagnosis
Hematology consultation was called for unexplained pancytopenia. Based on iron panel results and increased values for CRP and ESR, the initial concern was for anemia of chronic disease. Suspicion for hemophagocytic lymphohistiocytosis (HLH) was low due to the absence of splenomegaly, further corroborated with resolving thrombocytopenia and ferritin levels during her admission [6]. Suspicion for adult-onset Still’s disease was extremely low due to no splenomegaly, rash, or reported history of recent sore throat. The absence of schistocytes on peripheral blood smear made thrombotic thrombocytopenic purpura (TTP) unlikely. Cryoglobulin screen was positive, with trace cryoprecipitates detected. Serum and urine protein electrophoresis was negative for monoclonal gammopathy. HFE gene mutation was negative.
Her repeat CT chest/abdomen/pelvis did not show lymphadenopathy at axillary, supraclavicular, mediastinal, hilar, retroperitoneal, mesenteric, or inguinal areas. She was transferred to the intensive care unit (ICU) after becoming tachycardic and significant metabolic acidosis with serum lactate elevating to 5.5 mmol/L. She was eventually intubated for ventilation support. Driven by the unexplained cytopenia, a sternal bone marrow aspiration and biopsy was performed that showed RS cells. There was no evidence for HLH. The cells stained positive for CD15, CD30, PAX5, EBV, and negative for CD20 (Fig. 2). A diagnosis of HL was established.
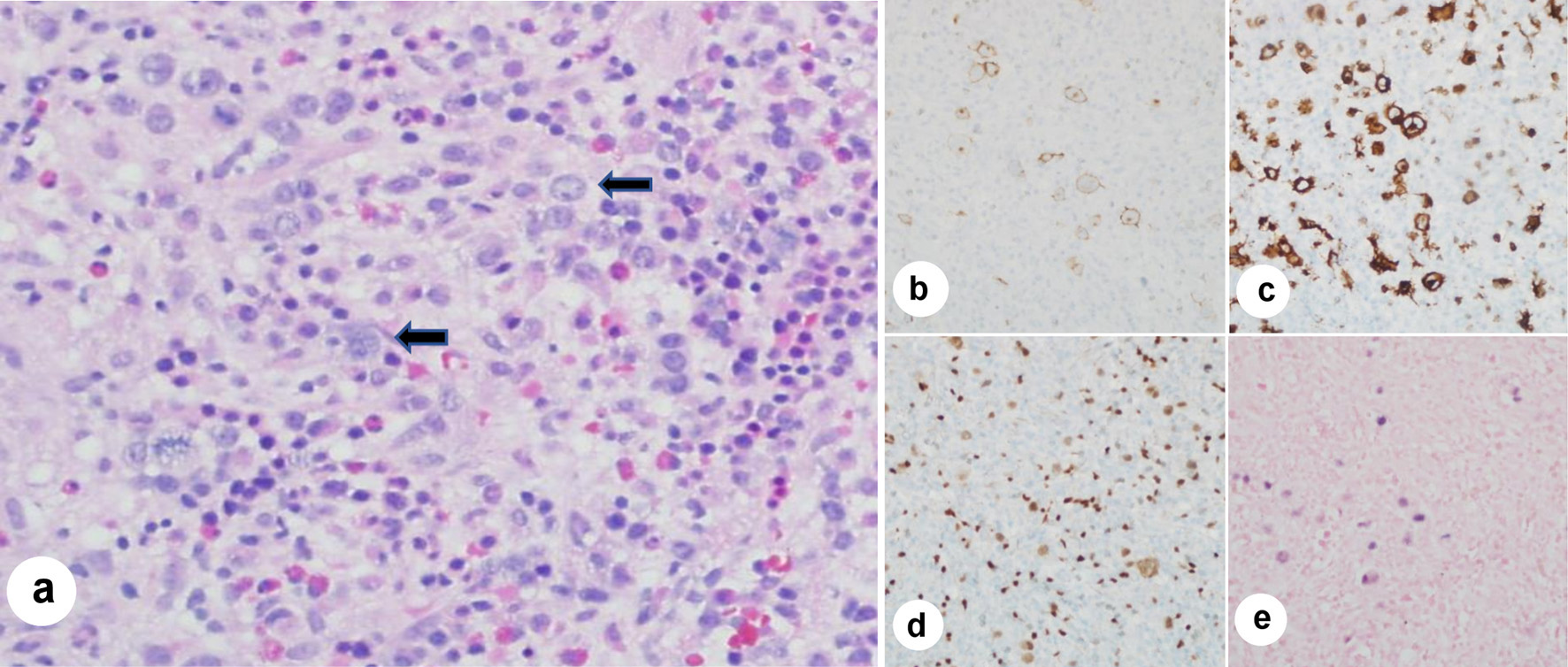 Click for large image | Figure 2. Bone marrow aspiration immunohistochemistry stains and in situ hybridization (ISH) stains. (a) Low power (× 20 magnification) hematoxylin and eosin stain of bone marrow aspirate clot shows marrow particles with Reed-Sternberg (RS) cells having prominent nucleoli and occasional bilobed forms (arrows) in a background of mixed mature and immature hematopoietic cells. (b) Photomicrograph (× 30 magnification) depicting CD 30+ immunohistochemistry (IHC) shows membranous and Golgi staining in RS cells. (c) Photomicrograph (× 30 magnification) depicting CD15+ IHC shows membranous and Golgi staining in the RS cells too, along with the staining of background granulocytes. (d) Photomicrograph (× 30 magnification) depicting PAX 5+ IHC shows positive staining in RS cells (lighter staining) and background B lymphocytes (darker staining). (e) Photomicrograph (× 30 magnification) depicting EBER ISH shows nuclear staining in RS cells, indicating Epstein-Barr virus (EBV) positivity (photomicrographs credit to Mary Hansen Smith, MD, Department of Pathology, Banner University Medical Center, Tucson, Arizona). |
Takotsubo cardiomyopathy complicated her disease course, resulting in a drop in her ejection fraction (EF) from 55% to 23%, along with the incidence of an apical left ventricle thrombus, warranting initiation of heparin drip. She also developed bilateral pleural effusions requiring thoracentesis, which revealed the fluid to be transudative. She was negative for HIV in the blood.
Treatment
A modified chemotherapy regimen of cyclophosphamide (750 mg intravenous (IV) for 1 day), vincristine (2 mg IV for 1 day), and dexamethasone (40 mg orally (PO) daily for 4 days) was given. Doxorubicin was avoided due to her reduced EF. Allopurinol 300 mg oral twice daily with adequate IV fluid hydration was proceeded. She was monitored for tumor lysis syndrome with daily labs, including uric acid, phosphorus, potassium, and calcium. Her repeat echocardiogram demonstrated an increase in EF to 59%.
Follow-up and outcomes
A plan was made to discharge the patient after a course of cytoreductive chemotherapy and start her on AVD (doxorubicin, vinblastine, dacarbazine) + BV (brentuximab vedotin) combination in the outpatient setting. However, her medical condition deteriorated quickly. She was admitted to ICU for hypotension associated with hyponatremia. The patient died of cardiovascular arrest a few days later without receiving another cycle of chemotherapy.
| Discussion | ▴Top |
HL may only present as lymphadenopathy or a mediastinal mass with or without the presence of constitutional symptoms. Some patients may only present with fatigue, fever, or pruritus symptoms. Obtaining a comprehensive history, including but not limited to the onset, duration, and extent of lymphadenopathy, constitutional symptoms, and possible family history of HL is vital for appropriately differential diagnosis of HL. A thorough and extensive physical examination is essential to evaluate the extent of HL involvement. Positron emission tomography (PET)/CT scan and other image study are required to stage the HL. A tissue biopsy is the gold standard to confirm and stratify the subtype of HL.
Stage IV HL is characterized by the extralymphatic organs involvement no matter whether there is lymphatic tissue involvement. EBV and HIV infections are prevalent in geographic regions with poor socioeconomic development and serve as risk factors for HL development. However, only a small fraction of EBV infected individuals develop EBV-positive HL. About 20-50% of HL cases in North America and Europe are EBV positive. Almost all cHL cases in tropical and developing countries are EBV positive [7].
Several case report series described diagnosis of PBMHL in HIV patients. These studies demonstrated that PBMHL in HIV patients present a more aggressive disease course with significantly shorter median survival time than typical HL in HIV patients [8, 9]. Ponzoni et al reported that the median survival time in HIV patients with PBMHL was 4 months, but in HIV patients with cHL it was 15 months [8]. Shah et al reported that the median age of HIV patients with PBMHL was 36 years old, which was significantly younger than HIV-negative PBMHL patients who were older than 50 years of age [9]. The atypical clinical presentation of PBMHL, at least partially, contributes to the delay of diagnosis, increased mortality rates, and shorter survival times. There have been reports of PBMHL in the HIV-negative pediatric patients, and these cases also carry a delayed time to diagnosis and a worse prognosis [10].
Due to the relatively unusual presentation, misdiagnosis and delayed treatment of PBMHL are commonly seen. Recent literature has suggested replacing bone marrow biopsy with PET/CT scans to detect bone marrow involvement in extranodal HL or PBMHL. However, false-negative cases can occur with PET/CT. Therefore, bone marrow biopsy continues to be the gold standard to confirm the involvement of the bone marrow in HL patients with PET/CT employed for follow-up. So far, there are only seven cases of PBMHL reported (Table 1) [10-15].
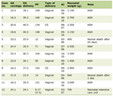 Click to view | Table 1. Reported Cases in Literatures of Isolated Bone Marrow Involvement in HIV-Negative Adults |
As outlined above, the current first-line combination chemotherapy for HL is ABVD. But there is scarcity of guidelines for PBMHL management. Due to this patient’s poor baseline status, treatment was adjusted to offer a better safety profile with cyclophosphamide, vincristine, and dexamethasone alone. A study conducted by the Nordic Lymphoma Group in elderly HL patients with comorbidities showed cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) to be an effective regimen with less associated toxicity [11].
Conclusions
This case report highlights the presence of cHL as an isolated bone marrow disorder in immunocompetent patients without HIV infection. Due to the atypical presentation of the disease, PBMHL patients are often misdiagnosed, leading to delays in management, which is further complicated by the disease’s aggressive nature. Therefore, an early bone marrow biopsy is warranted in patients with fever of unknown origin, unexplained cytopenia, and a rapidly worsening clinical course. HL usually has a fairly good curative rate after treating with proper chemotherapy regimens, but the prognosis of PBMHL continues to be poor. ECHELON-1 trial studying the efficacy of adding the antibody-drug conjugate brentuximab to the existing chemotherapy regimen of AVD for advanced HL showed robust and durable improvement in progression-free survival versus ABVD [12]. It is worth to try this innovative combination regimen on PBMHL.
Learning points
The mentioned report highlights a case of cHL involving the bone marrow primarily in an immunocompetent patient. Identifying PBMHL early can result in timely initiation of chemotherapy.
Acknowledgments
Photomicrographs are credited to Mary Hansen Smith MD, Department of Pathology, Banner University Medical Center, Tucson, Arizona.
Financial Disclosure
This study was supported by: AA&MDSIF research grant to JJP (146818), American Cancer Society grant to JJP (124171-IRG-13-043-02), JTTai&Co Foundation Cancer research grant to JJP, NIH/NCI grant to JJP (P30CA023074), and a University of Arizona Cancer Center research grant to JJP.
Conflict of Interest
The authors declare that they have no conflict of interest to disclose.
Informed Consent
Informed consent was obtained from the individual participant included in this study.
Author Contributions
AD and JJP initiated the research idea and concept. AD treated this patient. JJP and AD designed this study and wrote this manuscript. VFE participated in manuscription formation.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Shimabukuro-Vornhagen A, Haverkamp H, Engert A, Balleisen L, Majunke P, Heil G, Eich HT, et al. Lymphocyte-rich classical Hodgkin's lymphoma: clinical presentation and treatment outcome in 100 patients treated within German Hodgkin's Study Group trials. J Clin Oncol. 2005;23(24):5739-5745.
doi pubmed - Mauch PM, Kalish LA, Kadin M, Coleman CN, Osteen R, Hellman S. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer. 1993;71(6):2062-2071.
doi - Adams HJ, Kwee TC, de Keizer B, Fijnheer R, de Klerk JM, Littooij AS, Nievelstein RA. Systematic review and meta-analysis on the diagnostic performance of FDG-PET/CT in detecting bone marrow involvement in newly diagnosed Hodgkin lymphoma: is bone marrow biopsy still necessary? Ann Oncol. 2014;25(5):921-927.
doi pubmed - Bonadonna G, Zucali R, Monfardini S, De Lena M, Uslenghi C. Combination chemotherapy of Hodgkin's disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer. 1975;36(1):252-259.
doi - Engert A, Diehl V, Franklin J, Lohri A, Dorken B, Ludwig WD, Koch P, et al. Escalated-dose BEACOPP in the treatment of patients with advanced-stage Hodgkin's lymphoma: 10 years of follow-up of the GHSG HD9 study. J Clin Oncol. 2009;27(27):4548-4554.
doi pubmed - Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131.
doi pubmed - Leoncini L, Spina D, Nyong'o A, Abinya O, Minacci C, Disanto A, De Luca F, et al. Neoplastic cells of Hodgkin's disease show differences in EBV expression between Kenya and Italy. Int J Cancer. 1996;65(6):781-784.
doi - Ponzoni M, Fumagalli L, Rossi G, Freschi M, Re A, Vigano MG, Guidoboni M, et al. Isolated bone marrow manifestation of HIV-associated Hodgkin lymphoma. Mod Pathol. 2002;15(12):1273-1278.
doi pubmed - Shah BK, Subramaniam S, Peace D, Garcia C. HIV-associated primary bone marrow Hodgkin's lymphoma: a distinct entity? J Clin Oncol. 2010;28(27):e459-460.
doi pubmed - Alkendi J, Renzi S, Manson D, Abdelhaleem M, Naqvi A, Punnett A. Hodgkin Lymphoma in an adolescent with isolated bone marrow and bone involvement: a case report and a review of the literature. J Pediatr Hematol Oncol. 2021;43(1):e115-e118.
doi pubmed - Kolstad A, Nome O, Delabie J, Lauritzsen GF, Fossa A, Holte H. Standard CHOP-21 as first line therapy for elderly patients with Hodgkin's lymphoma. Leuk Lymphoma. 2007;48(3):570-576.
doi pubmed - Straus DJ, Dlugosz-Danecka M, Connors JM, Alekseev S, Illes A, Picardi M, Lech-Maranda E, et al. Brentuximab vedotin with chemotherapy for stage III or IV classical Hodgkin lymphoma (ECHELON-1): 5-year update of an international, open-label, randomised, phase 3 trial. Lancet Haematol. 2021;8(6):e410-e421.
doi - Morita Y, Emoto M, Serizawa K, Rai S, Hirase C, Kanai Y, Ohyama Y, et al. HIV-negative primary bone marrow Hodgkin lymphoma manifesting with a high fever associated with hemophagocytosis as the initial symptom: a case report and review of the previous literature. Intern Med. 2015;54(11):1393-1396.
doi pubmed - Hoti M, Sadiku S, Ukimeraj A, et al. HL-243: unusual primary bone marrow Hodgkin lymphoma presentation-epidemiological data from the University Clinical Center of Kosovo between 2009-2020. Clinical Lymphoma Myeloma and Leukemia. 2021;21:S371-S372.
doi - Lamba B, Talreja VT, Sudeep V, Mittal S. A case report of primary Hodgkin's disease of bone marrow. Sahel Medical Journal. 2015;18(2):87.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.