

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 11, Number 8, August 2020, pages 256-261
Adult Lymphoma-Associated Hemophagocytic Lymphohistiocytosis: A Clinical Case Series in a Predominantly Hispanic Cohort
Amrita Desaia, e, f, Eduardo E. Saulb, e, Jennifer R. Chapmanc, Lazaros Lekakisd, Agustin Pimenteld
aDepartment of Hematology-Oncology, OHSU Knight Cancer Institute, Oregon Health Sciences University, OR, USA
bDepartment of Medicine, University of Miami Miller School of Medicine/Jackson Memorial Hospital, Miami, FL, USA
cDepartment of Pathology and Laboratory Medicine, University of Miami Miller School of Medicine, Miami, FL, USA
dDivision of Hematology-Oncology, Department of Medicine, Sylvester Comprehensive Cancer Center, University of Miami, Miami, FL, USA
eThese authors contributed equally to this work.
fCorresponding Author: Amrita Desai, Department of Hematology-Oncology, OHSU Knight Cancer Institute, Oregon Health Sciences University, 1095 Exchange St, Astoria, OR 97103, USA
Manuscript submitted June 5, 2020, accepted June 23, 2020, published online July 21, 2020
Short title: Adult LA-HLH Case Series: A Hispanic Cohort
doi: https://doi.org/10.14740/jmc3521
| Abstract | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a systemic inflammation disorder secondary to immune dysregulation. Patients may present with fevers, splenomegaly, bone marrow failure and hemophagocytosis, among other clinical and laboratory findings. Lymphoma-associated HLH (LA-HLH) is a puzzling diagnosis given both conditions overlapping presentation. There are currently no established treatment guidelines for LA-HLH. We conducted a retrospective search of the tumor registry and pathology database at the University of Miami/Jackson Memorial Hospital using Pathology Laboratory Information System (LIS) and natural language search. We identified adult patients with a combined diagnosis of lymphoma and HLH between January 2008 and July 2018. Data from nine LA-HLH patients were identified and reviewed. The median age was 53 years (range 19 - 73), with 78% of cases of Hispanic origin. Lymphoma subtypes consisted of six T-cell/NK-cell neoplasms: two peripheral T-cell lymphoma (PTCL), not otherwise specified (NOS); two Epstein-Barr virus (EBV)+ extranodal NK-/T-cell lymphomas; one EBV+, CD8+, PTCL, NOS; one EBV+, post-transplant lymphoproliferative disorder-anaplastic large cell lymphoma, anaplastic lymphoma kinase negative (PTLD ALCL ALK-); and three B-cell neoplasms: one EBV+ diffuse large B-cell lymphoma (DLBCL); two DLBCL, NOS. HLH and lymphoma were diagnosed simultaneously in six out of nine cases. Hemophagocytosis phenomena were demonstrated in seven out of nine cases. Treatment consisted of combined HLH and lymphoma therapies in four cases, while lymphoma-directed therapy was applied to four patients; another case was treated with a modified version of the HLH-1994 protocol. Overall, a total of five cases were exposed to HLH-directed regimens (HLH-1994/2004). Three patients had refractory LA-HLH and entered hospice care, whereas another three cases succumbed to treatment-related complications. Of the seven cases that were evaluable for lymphoma response, four cases (57%) achieved complete response (CR), and three of them (43%) were alive with no evidence of recurrence at 10, 16 and 52 months as of the last contact. Herein, we describe our unique experience of an LA-HLH case series in a predominantly Hispanic population in South Florida. The diagnosis is challenging, often delayed, and the prognosis is dismal in refractory cases despite currently available rescue therapies. Furthermore, we describe for the first time the association between HLH and PTLD ALCL.
Keywords: Lymphoma; Hemophagocytic lymphohistiocytosis; Phagocytosis; NK cell; Hyperferritinemia; HLH-200
| Introduction | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) [1, 2] is a histiocytic disorder characterized by extreme immune deregulation and severe systemic inflammation. Patients commonly present with a systemic inflammatory response syndrome (SIRS) characterized by protracted fevers, bone marrow failure, splenomegaly and hemophagocytosis. Liver dysfunctions manifested as elevated transaminases and bilirubin are often present and can be life-threatening. Elevated ferritin, sometimes above 10,000 ng/mL, and depressed natural killer (NK) cell function are consistent features of HLH; however, the most specific parameter used to establish the diagnosis and follow disease activity is the serum soluble interleukin-2 receptor/CD25 (sCD25). Hemophagocytosis, although present and frequently pronounced in HLH, is not pathognomonic; infections, autoimmune disorders, lymphomas (especially of T-cell subtypes) and leukemias can be associated with hemophagocytosis phenomena without showing other HLH features [1, 2].
Although HLH is traditionally classified as hereditary or acquired, a genetic defect is currently believed to increase the risk of acquiring HLH after certain triggers. PRF1 gene, first described in the pathogenesis and accounting for 20-40% of hereditary HLH, encodes for perforin, a pore-forming protein with a critical role in the cytotoxic function of NK cells and CD8+ T cells. Other gene mutations linked to familial HLH include UNC13D, STX11, STXBP2, Rab27a, SH2D1A, or BIRC4. Homozygosity or compound heterozygosity for these mutations commonly leads to HLH during the first 2 years of life. However, simple heterozygosity may predispose adults to HLH after specific triggers such as viral infections (Epstein-Barr (EBV), cytomegalovirus (CMV), human herpesvirus-6 (HHV-6) and others), malignancies (lymphoproliferative disorders including B- and T-cell lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia (AML) with t(8;16) and rarely germ cell tumors or Ewing sarcoma), immunodeficiencies (HIV/AIDS, X-linked lymphoproliferative disorders, Griscelli and Chediak-Higashi syndromes), autoimmune diseases (juvenile rheumatoid arthritis and adult Still’s disease) and solid organ or allogeneic stem cell transplantation (allo-SCT) [1-3].
The underlying pathophysiology in HLH is the failure of homeostatic mechanisms to suppress activated macrophages, which abnormally propagate systemic inflammation and tissue damage through mechanisms of cytokine release (interleukin-6, tumor necrosis factor-α and interferon-γ) and uncontrolled phagocytosis. The unrestrained macrophage activity also leads to a state of moderate immunodeficiency with consequences of increased rates of infections and malignancies [1-3].
According to the 2004 HLH guidelines, the diagnosis of HLH requires either the identification of one of the HLH mutations or fulfillment of five out of the following eight diagnostic criteria (clinical, laboratory and histopathologic): fevers, splenomegaly, cytopenias of at least two lineages, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytic activity, low-absent NK-cell activity, hyperferritinemia (> 500 µg/L) and elevated sCD25 (> 2,400 U/mL) [4, 5].
HLH usually evolves into a rapidly fatal disease if left untreated. Based on two landmark studies performed mainly in the pediatric HLH population (HLH-1994/HLH-2004 protocols), the current treatment of HLH is a multidrug regimen comprising dexamethasone, etoposide and cyclosporine. In cases with central nervous system (CNS) disease or progressive neurological symptoms, intrathecal methotrexate is indicated [4]. Allo-SCT may be considered for patients with familial, persistent, or recurrent disease [1].
Lymphoma-associated HLH (LA-HLH) is a rare disorder carrying grave prognosis. Both T- and B-cell lymphomas can cause uncontrolled systemic responses and be the trigger for secondary HLH. Factors including the complexity and overlap in their clinical manifestations can lead to a delay in the identification and recognition of both entities. Currently, there are no established treatment guidelines for LA-HLH [2, 5].
| Case Reports | ▴Top |
We conducted a retrospective search of the tumor registry and pathology database at the University of Miami/Jackson Memorial Hospital to identify adult patients with the combined diagnosis of lymphoma and HLH between January 2008 and July 2018, using Pathology Laboratory Information System (LIS) and natural language search. Data from nine identified LA-HLH patients were reviewed to gain further understanding of the clinical features and treatment outcomes of our adult population of patients with LA-HLH. This study was reviewed and approved by the Institutional Review Board of both institutions.
The main clinical characteristics of our retrospective cohort are summarized in Table 1 and molecular analysis results are summarized in Table 2. While most series demonstrated a male prevalence [6, 7], our series has a female predominance (56%) similar to Yu et al [8]. The median age of our cohort is 53 years (range 19 - 73), with 78% of cases of Hispanic origin corresponding to our geographic location. Lymphoma subtypes consisted of six T-cell/NK-cell neoplasms (67%): two peripheral T-cell lymphoma (PTCL), not otherwise specified (NOS); two EBV+ extranodal NK-/T-cell lymphomas; one EBV+, CD8+, PTCL, NOS; one EBV+, post-transplant lymphoproliferative disorder-anaplastic large cell lymphoma, anaplastic lymphoma kinase negative (PTLD ALCL ALK-); and three B-cell neoplasms (33%): one EBV+ diffuse large B-cell lymphoma (DLBCL); two DLBCL, NOS. The clinical manifestations observed across all our nine cases included fevers, constitutional symptoms, splenomegaly and cytopenias. In seven out of the nine cases, hemophagocytosis phenomena were established in tissue specimens collected either from the bone marrow or the affected organs. In most of the cases (except cases 1, 3 and 4), HLH and lymphoma were diagnosed simultaneously. In case 1, HLH was diagnosed concurrently with second lymphoma relapse, 36 months after initial lymphoma diagnosis. In case 3, HLH preceded the lymphoma diagnosis by a period of 12 weeks, while in case 4, HLH arose 13 weeks following the diagnosis of PTLD ALCL ALK-, these occurring 5 years following a kidney transplant. In our cohort, five out of the nine cases (56%) tested positive on Epstein-Barr encoding region in situ hybridization (EBER-ISH) analysis, comparable to the series of Cattaneo et al and Han et al, reporting rates of 41% and 68% EBER-ISH positivity, respectively [9, 10]. In addition, the association between adult HLH and PTLD ALCL ALK- is not previously described in the literature [6-13].
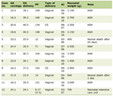 Click to view | Table 1. Clinical Features and Outcomes |
Click to view | Table 2. Molecular Analysis |
In our series, four cases received the combination of HLH plus lymphoma therapies, four cases had lymphoma-directed therapy and one case was treated with a modified version of the HLH-1994 protocol. Overall, a total of five cases received the HLH regimens (HLH-1994/2004). The selected specific lymphoma therapies for each patient in this case series are illustrated in Table 1. Three patients had refractory LA-HLH and entered hospice care. Two of them developed sepsis-related complications during the course of treatment leading to multiorgan failure and subsequent death, and a third patient succumbed 13 days after allo-SCT. Of the seven cases that were evaluable for lymphoma response (cases 1, 3, 4, 5, 6, 8 and 9), four (57%) of them achieved complete remission (CR), and three cases (43%) were refractory to therapy. Three out of the four cases that responded to treatment (CR) were alive with no evidence of recurrence at 10, 16 and 52 months as of last time contact.
| Discussion | ▴Top |
LA-HLH is a rare entity described in the medical literature to have an incidence rate of 0.9%, as illustrated by Machaczka and colleagues [14]. Supplementary Material 1 (www.journalmc.org) summarizes the published series analyzing the characteristics and outcomes of LA-HLH. Even though our series is built on a demographic of mostly Hispanics, we did not find significant clinical differences in their disease manifestations and treatment outcomes as compared to the reviewed published series. Our experience in LA-HLH indicates that NK-/T-cell lymphomas constitute the most common associated lymphoid malignancy, which correlates with the up-to-date literature. The mean survival of our LA-HLH patient group is 356 days, whereas the survival of the T-cell lymphomas is found to be higher compared to the B-cell lymphoma subgroup; however, this is just an observation given the limitation of a small descriptive study.
In 2015, the Study Group on HLH Subtypes of the Histiocyte Society published consensus recommendations for the management of malignancy-associated HLH (MA-HLH) [5]. Due to the lack of prospective trials, there is no explicit agreement on whether patients should receive HLH-directed therapy (dexamethasone, etoposide and cyclosporine), malignancy-focused interventions, or a combination of both. The conventional HLH protocols aimed at suppressing the overactive immune system have shown to improve survival in HLH cases. While treating the underlying malignancy seems natural, a 2017 consensus review from Daver et al [2] noted higher mortality rates when lymphoma-directed therapies were used alone, leading to a recommendation of a two-step approach: treating first the cytokine storm and T-cell proliferation with etoposide, corticosteroids, anti-thymocyte globulin and immunoglobulins followed by a lymphoma-directed therapy. Hence, merging HLH- and cancer-directed therapies, for instance, adding etoposide to the frequently used lymphoma regimens such as cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP), or initiating HLH-directed therapy as a first step to gain control of the life-threatening HLH cytokine storm syndrome followed by malignancy-focused treatment are both acceptable strategies [2, 5]. Another suggestion from the Study Group is to add anti-B-cell therapy such as rituximab in EBV-positive lymphomas and consider allo-SCT consolidation for LA-HLH, which is linked to improved survival outcomes [2, 5]. Additionally, owing to the severe immunosuppressive state and high incidences of infectious complications, supportive measures such as anti-bacterial, anti-fungal, anti-viral and pneumocystis prophylaxis along with surveillance for secondary viral infections and reactivations (CMV, EBV and fungi) should be strongly advocated in the treatment plan. Finally, treatment should be decided on a case-by-case basis, contemplating each patient’s clinical condition, co-morbidities, underlying malignancy and potential drug toxicities [1, 2, 5]. Relapsed or refractory patients may be rescued with DEP regimen (doxorubicin, etoposide and methylprednisolone) as per a Chinese report [2]. Similarly, several case-reports/case-series have shown alemtuzumab (anti-CD52 monoclonal antibody) and anakinra (interleukin-1 receptor antagonist) to be effective salvage therapies [1, 2, 5]. Recently, the FDA approved emapalumab (anti-interferon-γ monoclonal antibody) based on results from a pivotal multicenter, open-label, single-arm trial for patients with refractory, recurrent or progressive disease, or intolerance with conventional HLH therapy [15]. Furthermore, ongoing trials are evaluating the use of the Janus kinase/signal transducer and activator of transcription (JAK-STAT) inhibitor ruxolitinib in HLH [2].
Conclusion
Herein, we describe our unique experience of an LA-HLH case series in a predominantly Hispanic population in South Florida. Furthermore, we describe for the first time the association between HLH and PTLD ALCL. LA-HLH is an aggressive disease with a complex clinical presentation. The diagnosis is challenging, often delayed, and prognosis is dismal in refractory scenarios despite the currently available rescue therapies. Future prospective studies focusing on early diagnosis, management and novel targeted molecules will hopefully further improve the outcomes of patients with LA-HLH.
| Supplementary Material | ▴Top |
Suppl 1. Literature Review.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
The manuscript has been sufficiently de-identified to protect the patients’ information. This study has been reviewed and approved by the University of Miami and Jackson Memorial Hospital IRB and granted waiver of consent under protocol number 20191229.
Author Contributions
All authors contributed to the editing of the manuscript. AD, EES and AP wrote the manuscript and made the accompanying tables. Each author has reviewed the final version of the manuscript and approves it for publication.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052.
doi pubmed - Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, Blechacz B, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123(17):3229-3240.
doi pubmed - Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood. 2013;121(26):5184-5191.
doi pubmed - Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131.
doi pubmed - Lehmberg K, Nichols KE, Henter JI, Girschikofsky M, Greenwood T, Jordan M, Kumar A, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica. 2015;100(8):997-1004.
- Jiang T, Ding X, Lu W. The prognostic significance of Beta2 microglobulin in patients with hemophagocytic lymphohistiocytosis. Dis Markers. 2016;2016:1523959.
doi pubmed - Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 2014;89(4):484-492.
doi pubmed - Yu JT, Wang CY, Yang Y, Wang RC, Chang KH, Hwang WL, Teng CL. Lymphoma-associated hemophagocytic lymphohistiocytosis: experience in adults from a single institution. Ann Hematol. 2013;92(11):1529-1536.
doi pubmed - Cattaneo C, Oberti M, Skert C, Passi A, Farina M, Re A, Tozzi P, et al. Adult onset hemophagocytic lymphohistiocytosis prognosis is affected by underlying disease and coexisting viral infection: analysis of a single institution series of 35 patients. Hematol Oncol. 2017;35(4):828-834.
doi pubmed - Han AR, Lee HR, Park BB, Hwang IG, Park S, Lee SC, Kim K, et al. Lymphoma-associated hemophagocytic syndrome: clinical features and treatment outcome. Ann Hematol. 2007;86(7):493-498.
doi pubmed - Li F, Li P, Zhang R, Yang G, Ji D, Huang X, Xu Q, et al. Identification of clinical features of lymphoma-associated hemophagocytic syndrome (LAHS): an analysis of 69 patients with hemophagocytic syndrome from a single-center in central region of China. Med Oncol. 2014;31(4):902.
doi pubmed - Aljitawi OS, Boone JM. Lymphoma-associated hemophagocytic lymphohistiocytosis. Blood. 2012;120(5):932.
doi pubmed - Fung KM, Chakrabarty JH, Kern WF, Magharyous H, Gehrs BC, Li S. Intravascular large B-cell lymphoma with hemophagocytic syndrome (Asian variant) in a Caucasian patient. Int J Clin Exp Pathol. 2012;5(5):448-454.
- Machaczka M, Vaktnas J, Klimkowska M, Hagglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52(4):613-619.
doi pubmed - Locatelli F, Jordan MB, Allen CE, Cesaro S, Rizzari C, Rao A, Degar B, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Blood. 2018;132(suppl 1):LBA-6
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.