

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 11, Number 5, May 2020, pages 115-119
Anesthetic Care of a Child With Merosin-Deficient Muscular Dystrophy
Dolly M. Munlemvoa, d, Anuradha Kanaparthib, Joseph D. Tobiasa, c
aDepartment of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, Columbus, OH, USA
bNortheast Ohio Medical University (NEOMED), Rootstown, OH, USA
cDepartment of Anesthesiology & Pain Medicine, The Ohio State University College of Medicine, Columbus, OH, USA
dCorresponding Author: Dolly M. Munlemvo, Department of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, 700 Children’s Drive, Columbus, OH 43205, USA
Manuscript submitted March 30, 2020, accepted April 7, 2020
Short title: Merosin-Deficient Muscular Dystrophy
doi: https://doi.org/10.14740/jmc3460
| Abstract | ▴Top |
Merosin-deficient congenital muscular dystrophy (MDCMD) is a progressive autosomal recessive disorder caused by the lack of expression of the α2-chain of laminin-211 glycoprotein. The defect results in skeletal muscle dysfunction with severe muscle weakness, hypotonia, proximal joint contractures, facial dysmorphism, and late or failed ambulation. Given the progressive neuromuscular involvement and the potential for neuromuscular scoliosis, patients frequently require anesthetic care during surgical procedures to correct orthopedic deformities. We present a 9-year-old girl with MDCMD who required anesthetic care during insertion of growing rods to correct neuromuscular scoliosis. Previous reports of anesthetic care for patients with MDCMD are presented and options for perioperative care are reviewed.
Keywords: Scoliosis; Merosin; Congenital muscular dystrophy; Laminin-α2
| Introduction | ▴Top |
Merosin-deficient congenital muscular dystrophy (MDCMD) is a progressive autosomal recessive disorder that results in the deficient production of the α2-chain of laminin (merosin), a protein of the extracellular matrix of skeletal muscle that is encoded by genes located on chromosome 6q22-23 [1, 2]. First described in 1994 by Tome et al, it is one of the most common forms of CMD [3]. The cellular defect results in the premature truncation, and thereby defective function of the laminin-211 glycoprotein, which plays a crucial role in establishing the structural and functional scaffolding between the basement membrane and the extracellular matrix in cardiac muscle, skeletal muscle, Schwann cells, oligodendrocytes and trophoblasts [1, 4-8]. The mechanical stress on muscle fibers without basement membrane support from laminin-211 leads to progressive muscle injury and atrophy, resulting in inflammation, scarring and elevation of creatinine phosphokinase (CPK) levels.
Clinical signs and symptoms include severe muscle weakness, hypotonia, proximal joint contractures, delayed and often unachieved ambulatory efforts and facial dysmorphism [8-12]. Muscle weakness, particularly of the intercostal and accessory muscles, predisposes patients to respiratory infections, which are the most common cause of death [1]. These clinical signs may be present at birth and generally manifest by 1 year of age. Central nervous system (CNS) and white matter abnormalities of the brain lead to intellectual disabilities and seizures [5, 10]. The diagnosis of MDCMD is confirmed by the clinical sign and symptoms, elevation of serum CPK levels, muscle biopsy showing abnormal basement membrane structure and gene analysis to determine LAMA2 mutations [1, 12].
Surgical intervention may be required in patients with MDCMD to correct neuromuscular and orthopedic deformities including kyphoscoliosis. We present a pediatric patient with MDCMD who required anesthetic care for a posterior spinal fusion secondary to neuromuscular scoliosis. Previous reports of anesthetic care for patients with MDCMD are presented and options for perioperative care are reviewed.
| Case Report | ▴Top |
Review of this case and presentation in this format is in accordance with the guidelines of the Institutional Review Board at Nationwide Children’s Hospital (Columbus, OH). The patient was a 9-year-old, 15-kg girl with MDCMD who presented for the insertion of growing rods to treat neuromuscular scoliosis of the thoracic region. She was diagnosed with MDCMD at 10 months of age, following a muscle biopsy and gene sequencing analysis. Significant past medical history included a birth injury to the brachial plexus, previous respiratory infections including left lower lobe pneumonia and rhinovirus infection as well as age-related osteoporosis with low bone density for age. Past surgical history included a diagnostic muscle biopsy at 10 months of age. Current medications included bisphosphonate infusions for osteoporosis, and as needed albuterol and saline aerosol treatments to facilitate clearance of respiratory secretions. Her physical examination demonstrated generalized hypotonia, scoliosis and contractures at the elbows, hips and knees. Preoperative echocardiogram and electrocardiogram were unremarkable. On the morning of the procedure, the patient was kept nil per os for 6 h. The patient was transported to the operating room where standard American Society of Anesthesiologists’ (ASA) monitors were placed. After breathing 70% nitrous oxide in oxygen, a 20-gauge peripheral intravenous cannula was placed. Anesthesia was induced with propofol (3 mg/kg) and fentanyl (1 µg/kg). After effective bag-valve-mask ventilation was demonstrated, neuromuscular blockade was provided by rocuronium (0.6 mg/kg). Direct laryngoscopy with a Wis-Hipple 1.5 blade revealed a Cormack-Lehane grade 2 view and a 5.0 cuffed endotracheal tube (ETT) was placed without difficulty. After anesthetic induction, a second peripheral intravenous cannula and a radial arterial cannula were placed using ultrasound guidance. Maintenance anesthesia included methadone (0.1 mg/kg) and total intravenous anesthesia (TIVA) with infusions of remifentanil and propofol. Intraoperative neurophysiological monitoring was performed with somatosensory and motor evoked potentials. The propofol infusion was titrated to maintain the bispectral index at 40 - 60 and the remifentanil was titrated to maintain the mean arterial pressure at baseline. To limit the need to allogeneic blood products, tranexamic acid (bolus of 50 mg/kg followed by an infusion at 5 mg/kg/h) and intraoperative blood salvage were used. Intraoperative hypotension was treated with intermittent doses of phenylephrine as needed. Submuscular MAGEC™ (Magnetic Expansion Control System) rods were placed with rib hooks at T5 through S2 with sacroiliac screws. Total intraoperative fluids included 200 mL of crystalloid, 450 mL of 5% albumin and 200 mL of packed red blood cells. The procedure lasted for approximately 6 h. Following the procedure, the patient was transported to the pediatric intensive care unit (PICU). When awake, her trachea was extubated to bilevel positive airway pressure (BiPAP) via a mask. The BiPAP was continued overnight and then intermittently for the first 48 postoperative hours. Postoperative analgesia was provided by intermittent intravenous acetaminophen and ketorolac administered around the clock for 48 h and nursing-controlled analgesia with hydromorphone. The remainder of the postoperative period was unremarkable and she was discharged to home on postoperative day 5.
| Discussion | ▴Top |
MDCMD is one of the congenital muscular dystrophies, a group of conditions that share early onset and a similar histologic appearance of the muscle. MDCMD typically presents with decreased in utero movement followed by hypotonia at birth. Differential diagnoses include other congenital neuromuscular disorders including peripheral neuropathy, limb-girdle muscular dystrophy, Emery-Dreifuss muscular dystrophy, central demyelination diseases, congenital myopathy and spinal muscular atrophy [1, 2, 5, 10]. The diagnosis is suggested by high serum CPK levels and a muscle biopsy showing the abnormal basement membrane structure. Definitive identification of the disorder is confirmed by gene sequencing analysis identifying the mutations in LAMA2. As there is no treatment for MDCMD, symptom management is tailored to correct the anatomical and physiologic consequences of the primary disease process and ameliorate the quality of life.
Anesthetic care begins with a thorough preoperative exam with identification of the end-organ involvement related to the primary disease process and associated comorbid conditions. In patients with MDCMD, primary anesthetic concerns include airway involvement as well as the respiratory and cardiovascular systems [13-15]. Airway management including bag-valve-mask ventilation and endotracheal intubation may be problematic in patients with facial dimorphism including micrognathia, midface hypoplasia, and other associated dysmorphic features as noted in our patient [16]. The potential for difficulties with airway management in patients with muscular dystrophy has been previously reported to be 3.4%, a percentage that is significantly higher than that routinely encountered in the pediatric population [17]. When there are concerns regarding airway management, spontaneous ventilation should be maintained until the airway is secured or adequate bag-valve-mask ventilation is demonstrated. The appropriate equipment for dealing with the difficult airway including indirect videolaryngoscopes should be readily available prior to anesthetic induction or airway management [18, 19]. Despite the dysmorphic features noted in our patient, airway management including bag-valve-mask ventilation and endotracheal intubation was not problematic.
The medications used for the induction and maintenance of anesthesia are guided by the primary disease process, its end-organ involvement and the need for intraoperative neurophysiological monitoring. A single case report has suggested the potential for malignant hyperthermia (MH) in patients with MDCMD [20]. Even if not specifically MH-sensitive, the prolonged administration of volatile anesthetic agents may result in destabilization of the sarcolemma with rhabdomyolysis and hyperkalemia [14]. Furthermore, maintenance anesthesia with TIVA may be preferred to facilitate intraoperative spinal cord monitoring with motor and somatosensory evoked potentials [21, 22]. Given these considerations, we chose to administer 50-70% nitrous oxide followed by placement of a peripheral intravenous cannula and intravenous induction. Additionally, as indicated, a topical anesthetic cream may also be placed preoperatively to provide cutaneous analgesia and facilitate peripheral venous cannulation.
An invariable finding with MDCMD is hypotonia with the potential involvement of upper airway and respiratory musculature. These issues may predispose these patients to upper airway obstruction or respiratory failure during the postoperative period. The risk of perioperative respiratory failure may be increased by pre-existing respiratory dysfunction from hypotonia, poor cough effort, chronic aspiration or recurrent pneumonia. Preoperative assessment of respiratory function using pulmonary function testing (PFT) is limited due to the cognitive involvement and age of these patients as PFT is cooperation and effort dependent [23, 24]. However, a room air oxygen saturation less than 95%, a history of recurrent pneumonia, or swallowing problems with aspiration may identify at risk patients. Preoperative preparation should include aggressive treatment of respiratory infections and as cognitive function permits, instruction regarding the use of techniques such as incentive spirometry during the postoperative period. Patients with pre-existing motor weakness and hypotonia may be sensitive to the effects of neuromuscular blocking agent (NMBAs), inhalational agents and sedative agents. The use of short-acting agents (remifentanil) and complete reversal of residual neuromuscular blockade (see below) is suggested. Per our routine for patients with preoperative respiratory comorbidities, we chose to extubate our patient’s trachea to non-invasive ventilation (BiPAP) to prevent atelectasis and allow for an easy transition to spontaneous ventilation [25, 26].
Muscular dystrophy and hypotonia have a significant impact on the selection, use and reversal of NMBAs during the perioperative period. Succinylcholine is absolutely contraindicated due to the risk of hyperkalemia [27, 28]. The spread of acetylcholine receptors over the entire sarcolemma leads to potassium efflux on depolarization following the administration of succinylcholine, resulting in potentially fatal arrhythmias related to hyperkalemia [27, 28]. Non-depolarizing NMBAs are safe in patients with various muscular dystrophies; however, a prolonged effect can be expected even with intermediate-acting agents such as atracurium, rocuronium or vecuronium [29]. Prior to their withdrawal, rapacuronium and mivacurium, two short-acting NMBAs, offered the potential option to achieve complete neuromuscular blockade for endotracheal intubation with an acceptable recovery time even for briefer procedures [30, 31]. More recently, the novel reversal agent for NMBAs, sugammadex, offers the potential to reverse even profound neuromuscular blockade in patients with neuromyopathic conditions [32].
As opposed to patients with Duchenne or Becker muscular dystrophy, there is limited information regarding the potential for and the magnitude of cardiac involvement in patients with MDCMD [33, 34]. As the expression of the laminin-α2 chain is particularly high in the heart, cardiac involvement may occur in a significant percentage of patients. Reported comorbid cardiac involvement has included conduction disturbances (right bundle branch block), dilated cardiomyopathy, systolic dysfunction, mitral valve regurgitation, pulmonary hypertension, arrhythmias and wall motion hypokinesis. Spyrou et al studied the cardiac function of a cohort of 16 children with CMD using echocardiography. Two of six merosin-deficient children had an ejection fraction less than 40%. The average ejection fraction of the merosin-deficient children was 43±11% compared to 53±5% in merosin-positive children [34]. Manifestations of cardiac involvement may occur later in the disease than the skeletal muscle hypotonia. Additionally, as reported by Jimenez et al, due to the extreme scoliosis and positioning required intraoperatively, hemodynamic compromise may occur related to anatomical obstruction of cardiac output [35]. Due to these concerns, preoperative echocardiography and electrocardiography are recommended to screen for comorbid cardiac involvement. If significant cardiac involvement is noted, preoperative consultation with pediatric cardiology may be indicated. Additionally, the presence of depressed myocardial function may impact the choice of agents for anesthetic induction as well as the need for invasive monitoring of central venous pressure.
Merosin is also expressed in the brain and magnetic resonance has demonstrated white matter involvement and other CNS involvement [36]. Microcephaly, mental retardation, cerebral atrophy, cortical dysplasia, white matter changes, pontine and cerebral atrophy or cerebral cyst can be found on brain magnetic resonance imaging (MRI). Clinical findings have included mental retardation, developmental delay and seizures [37, 38]. Preoperative identification of CNS involvement, optimization of preoperative seizure control and continuation of anti-epileptic medications when necessary during the perioperative period are required in these patients.
To date, we identified only four published reports involving a total of 19 anesthetics in 13 patients regarding the anesthetic care of patients with MDCMD (Table 1) [13, 20, 35, 39]. Aside from concerns for MH in one patient, the remainder of the perioperative care was uneventful. Fourteen anesthetic encounters included a volatile-based anesthetic and five TIVA. When caring for patients with MDCMD, perioperative concerns include potential difficulties with airway management and endotracheal intubation, cardiac involvement, hypotonia with a risk for postoperative respiratory insufficiency or failure and CNS involvement including seizures and developmental delay.
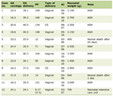 Click to view | Table 1. Reports of Anesthetic Care in Patients With Merosin-Deficient Congenital Muscular Dystrophy |
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
In accordance with IRB of Nationwide Children’s Hospital, need for documentation of informed consent waved.
Author Contributions
Dolly M. Munlemvo contributed to preparation of secondary and final drafts. Anuradha Kanaparthi contributed to preparation of initial drafts, review of final manuscript. Joseph Tobias contributed to concept, review of all drafts.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Nguyen Q, Lim KRQ, Yokota T. Current understanding and treatment of cardiac and skeletal muscle pathology in laminin-alpha2 chain-deficient congenital muscular dystrophy. Appl Clin Genet. 2019;12:113-130.
doi pubmed - Mohassel P, Foley AR, Bonnemann CG. Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol. 2018;71-72:188-204.
doi pubmed - Tome FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B, Barois A, et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. 1994;317(4):351-357.
- Turner C, Mein R, Sharpe C, Love DR. Merosin-deficient congenital muscular dystrophy: A novel homozygous mutation in the laminin-2 gene. J Clin Neurosci. 2015;22(12):1983-1985.
doi pubmed - Chan SH, Foley AR, Phadke R, Mathew AA, Pitt M, Sewry C, Muntoni F. Limb girdle muscular dystrophy due to LAMA2 mutations: diagnostic difficulties due to associated peripheral neuropathy. Neuromuscul Disord. 2014;24(8):677-683.
doi pubmed - Gawlik KI, Harandi VM, Cheong RY, Petersen A, Durbeej M. Laminin alpha1 reduces muscular dystrophy in dy(2J) mice. Matrix Biol. 2018;70:36-49.
doi pubmed - Yurchenco PD, McKee KK, Reinhard JR, Ruegg MA. Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Matrix Biol. 2018;71-72:174-187.
doi pubmed - He Z, Luo X, Liang L, Li P, Li D, Zhe M. Merosin-deficient congenital muscular dystrophy type 1A: A case report. Exp Ther Med. 2013;6(5):1233-1236.
doi pubmed - Fontes-Oliveira CC, Steinz M, Schneiderat P, Mulder H, Durbeej M. Bioenergetic impairment in congenital muscular dystrophy type 1A and leigh syndrome muscle cells. Sci Rep. 2017;7:45272.
doi pubmed - Nelson I, Stojkovic T, Allamand V, Leturcq F, Becane HM, Babuty D, Toutain A, et al. Laminin alpha2 Deficiency-Related Muscular Dystrophy Mimicking Emery-Dreifuss and Collagen VI related Diseases. J Neuromuscul Dis. 2015;2(3):229-240.
doi pubmed - Gavassini BF, Carboni N, Nielsen JE, Danielsen ER, Thomsen C, Svenstrup K, Bello L, et al. Clinical and molecular characterization of limb-girdle muscular dystrophy due to LAMA2 mutations. Muscle Nerve. 2011;44(5):703-709.
doi pubmed - Kim HJ, Choi YC, Park HJ, Lee YM, Kim HD, Lee JS, Kang HC. Congenital muscular dystrophy type 1A with residual merosin expression. Korean J Pediatr. 2014;57(3):149-152.
doi pubmed - Scrivener TA, Ross SM, Street NE, Webster RI, De Lima JC. A case series of general anesthesia in children with laminin alpha2 (merosin)-deficient congenital muscular dystrophy. Paediatr Anaesth. 2014;24(4):464-465.
doi pubmed - Cripe LH, Tobias JD. Cardiac considerations in the operative management of the patient with Duchenne or Becker muscular dystrophy. Paediatr Anaesth. 2013;23(9):777-784.
doi pubmed - Petersen CT, Norregaard FO. [Duchenne muscular dystrophy and anesthesia. A retrospective study]. Ugeskr Laeger. 1996;158(44):6274-6276.
- Butler MG, Hayes BG, Hathaway MM, Begleiter ML. Specific genetic diseases at risk for sedation/anesthesia complications. Anesth Analg. 2000;91(4):837-855.
doi pubmed - Muenster T, Mueller C, Forst J, Huber H, Schmitt HJ. Anaesthetic management in patients with Duchenne muscular dystrophy undergoing orthopaedic surgery: a review of 232 cases. Eur J Anaesthesiol. 2012;29(10):489-494.
doi pubmed - Engelhardt T, Weiss M. A child with a difficult airway: what do I do next? Curr Opin Anaesthesiol. 2012;25(3):326-332.
doi pubmed - Bryant J, Krishna SG, Tobias JD. The difficult airway in pediatrics. Adv Anesth. 2013;3:31-60.
doi - Shukry M, Guruli ZV, Ramadhyani U. Suspected malignant hyperthermia in a child with laminin alpha2 (merosin) deficiency in the absence of a triggering agent. Paediatr Anaesth. 2006;16(4):462-465.
doi pubmed - Martin DP, Bhalla T, Thung A, Rice J, Beebe A, Samora W, Klamar J, et al. A preliminary study of volatile agents or total intravenous anesthesia for neurophysiological monitoring during posterior spinal fusion in adolescents with idiopathic scoliosis. Spine (Phila Pa 1976). 2014;39(22):E1318-1324.
doi pubmed - Ravussin P, de Tribolet N, Wilder-Smith OH. Total intravenous anesthesia is best for neurological surgery. J Neurosurg Anesthesiol. 1994;6(4):285-289.
doi pubmed - Birnkrant DJ. The American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Pediatrics. 2009;(Suppl 4):S242-244.
doi pubmed - Bach JR, Ishikawa Y, Kim H. Prevention of pulmonary morbidity for patients with Duchenne muscular dystrophy. Chest. 1997;112(4):1024-1028.
doi pubmed - Jaber S, Michelet P, Chanques G. Role of non-invasive ventilation (NIV) in the perioperative period. Best Pract Res Clin Anaesthesiol. 2010;24(2):253-265.
doi pubmed - Pelosi P, Jaber S. Noninvasive respiratory support in the perioperative period. Curr Opin Anaesthesiol. 2010;23(2):233-238.
doi pubmed - Martyn JA, Richtsfeld M. Succinylcholine-induced hyperkalemia in acquired pathologic states: etiologic factors and molecular mechanisms. Anesthesiology. 2006;104(1):158-169.
doi pubmed - Kendig JJ, Bunker JP, Endow S. Succinylcholine-induced hyperkalemia: effects of succinylcholine on resting potentials and electrolyte distributions in normal and denervated muscle. Anesthesiology. 1972;36(2):132-137.
doi pubmed - Sethna NF, Rockoff MA, Worthen HM, Rosnow JM. Anesthesia-related complications in children with Duchenne muscular dystrophy. Anesthesiology. 1988;68(3):462-465.
doi pubmed - Frankowski GA, Johnson JO, Tobias JD. Rapacuronium administration to two children with Duchenne's muscular dystrophy. Anesth Analg. 2000;91(1):27-28.
doi pubmed - Tobias JD, Uslu M. Mivacurium administration in children with Duchenne muscular dystrophy. Anesth Analg. 2000;90(2):498-499.
doi - Tobias JD. Current evidence for the use of sugammadex in children. Paediatr Anaesth. 2017;27(2):118-125.
doi pubmed - Finsterer J, Ramaciotti C, Wang CH, Wahbi K, Rosenthal D, Duboc D, Melacini P. Cardiac findings in congenital muscular dystrophies. Pediatrics. 2010;126(3):538-545.
doi pubmed - Spyrou N, Philpot J, Foale R, Camici PG, Muntoni F. Evidence of left ventricular dysfunction in children with merosin-deficient congenital muscular dystrophy. Am Heart J. 1998;136(3):474-476.
doi - Jimenez N, Song K, Lynn AM. Hemodynamic instability during prone spine surgery in a patient with merosin-deficient congenital muscular dystrophy. Paediatr Anaesth. 2013;23(3):294-296.
doi pubmed - Ibrahim Abdulla JK, Vattoth S, Al Tawari AA, Pandey T, Abubacker S. Brain MRI features of merosin-negative congenital muscular dystrophy. Australas Radiol. 2007;51(Suppl):B221-223.
doi pubmed - Jones KJ, Morgan G, Johnston H, Tobias V, Ouvrier RA, Wilkinson I, North KN. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet. 2001;38(10):649-657.
doi pubmed - Xiong H, Tan D, Wang S, Song S, Yang H, Gao K, Liu A, et al. Genotype/phenotype analysis in Chinese laminin-alpha2 deficient congenital muscular dystrophy patients. Clin Genet. 2015;87(3):233-243.
doi pubmed - Pregardien C, Pirotte T, Veyckemans F. Pressure-support ventilation in a child with merosin-deficient congenital muscular dystrophy under sevoflurane anesthesia. Acta Anaesthesiol Belg. 2016;67(3):139-141.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.