

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 7, Number 5, May 2016, pages 191-194
Proliferative Glomerulonephritis With Monoclonal IgG Deposits in a Young Patient: A Case Report
Lara El Khourya, b, Suzanne El Sayegha, Theodore Strangea, Elie El Charabatya, Talal El Imada
aStaten Island University Hospital, NY, USA
bCorresponding Author: Lara El Khoury, Staten Island University Hospital, NY, USA
Manuscript accepted for publication February 16, 2016
Short title: Proliferative Glomerulonephritis
doi: http://dx.doi.org/10.14740/jmc2444w
| Abstract | ▴Top |
Proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) is a newly described entity that belongs to the class of disorders known as monoclonal gammopathy of renal significance (MGRS). The renal deposit in PGNMID is composed of monoclonal immunoglobulin (Ig). Microscopic findings typically show a non-fibrillar or non-microtubular pattern of Ig deposit without any organized structure. This entity affects adults and most patients present with nephrotic-range proteinuria and hematuria with or without renal failure. We report a unique case of PGNMID occurring in a young patient following a viral infection. The patient had significant proteinuria. There were no monoclonal proteins found in the serum. The renal biopsy was consistent with proliferative glomerulonephritis (GN) with monoclonal immune deposits. PGNMID is a novel form of GN that mimics immune-complex type GN on light and electron microscopy; thus, renal biopsy with careful attention to light-chain and IgG isotype staining is essential for diagnosis.
Keywords: Monoclonal immune deposits; Proliferative glomerulonephritis; Immunofluorescence
| Introduction | ▴Top |
Proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) is a rare entity caused by monoclonal deposition of IgG. PGNMID has similarities to immune complex glomerulonephritis (GN) on light and electron microscopy (EM); however, by immunofluorescence (IF), the glomerular deposits are monoclonal, staining for a single light-chain isotype and a single γ heavy-chain subclass, most commonly IgG3κ. This entity affects adults and most patients present with nephrotic-range proteinuria and hematuria with or without renal failure.
We report a case of a young healthy female patient who presented with proteinuria and a normal kidney function and was found to have monoclonal immune deposits on the kidney biopsy.
| Case Report | ▴Top |
A 20-year-old young female patient with no significant past medical history presented to the office for evaluation of worsening bilateral lower extremity edema and a 32-lb weight gain over the past 2 weeks. The patient reported a viral prodrome consisting of fever and sore throat that started 2 weeks ago. She denied any dysuria or hematuria but complained of facial swelling.
Vital signs revealed blood pressure of 120/70 mm Hg, hear rate of 72 beats/min, respiratory rate of 16/min, afebrile. Lung and heart examination was normal and there was no palpable cervical, supraclavicular, axillary, or inguinal lymphadenopathy. Abdominal exam was significant for hepatomegaly. Bilateral, three plus pitting lower extremity edema was present.
Her laboratory evaluation showed hemoglobin (Hb) of 12.3 g/dL, total leukocyte count of 17,250/mm3 (85% granulocytes), platelets of 378,000/mm3, and erythrocyte sedimentation rate (ESR) of 27 mm/h. Urine analysis showed protein of 100 mg/dL, red blood cell (RBC) of 0 - 3/hpf, and white blood cell (WBC) of 3 - 6/hpf. Spot urine protein/creatinine ratio was 1,853.28 mg/g CRE, blood urea was 17 mg/dL, serum creatinine was 0.8 mg/dL, Na/K/Cl was 137/5.4/108 mEq/L, Ca/P was 8.5 mg/5.6 mg/dL, serum protein was 4.5 g/dL, and albumin was 2 g/dL. Her serum complements (C3 and C4) were normal and serum cryoglobulin was initially positive but turned out to be negative on repeated testing. Rheumatoid factor, anti-nuclear antibody (ANA), and anti-glomerular basement membrane were normal. Serology tests for hepatitis A, hepatitis B, hepatitis C and human immunodeficiency virus were negative. Mycoplasma antibody IgG was negative; IgM was 322. Quantitative Strep O was negative. CMV IgG and IgM were negative. EBV IgG was positive, and IgM was negative. Serum protein electrophoresis (SPEP) and free light-chain assay were normal. Urine immunoelectrophoresis (UPEP) showed a total protein of 3,287 with no monoclonal proteins on immunofixation. On retroperitoneal ultrasound, kidneys were of normal size and echogenicity. Renal biopsy showed eight glomeruli with endocapillary proliferation and neutrophil infiltration but no crescent formation. Ultrastructurally, there were segmental to global subepithelial hump-shaped immune-type electron dense deposits with no associated GBM reaction or spikes. Segmental global mesangial deposits and segmental subendothelial deposits are also seen. Moderate podocyte effacement is noted. These findings were consistent with diffuse proliferative GN with exudates features and IgG3 kappa deposits.
| Discussion | ▴Top |
Dysproteinemia is characterized by excessive synthesis of Ig molecules or subunits, which may form paraprotein tissue deposits and produce an array of pathologic lesions, most commonly amyloidosis and monoclonal Ig deposition disease. Several renal diseases are associated with dysproteinemias, among which those manifesting monoclonal glomerular deposits of IgG are relatively uncommon. Glomerular diseases associated with monoclonal gammopathies include multiple myeloma (MM), monoclonal Ig deposition disease, type I cryoglobulinemic GN and amyloidosis. Of these, MM is the most commonly associated.
In 2004, Nasr et al reported 10 patients with an unusual form of glomerular injury caused by monoclonal IgG deposition that does not belong to a specific entity, which was termed PGNMID [1]. On IF, the glomerular deposits were monoclonal, staining for a single light-chain isotype and a single γ heavy-chain subclass. Light microscopy (LM) exhibited endocapillary proliferative or membranoproliferative GN, and EM revealed granular electron-dense deposits, mimicking ordinary immune-complex GN. Clinical presentations included proteinuria in 100% of patients (mean 24-h urine protein 5.8 g), renal insufficiency in 80% (mean serum creatinine 2.8 mg/dL), and microhematuria in 60%. A monoclonal serum or urine protein was identified in 50% of patients, although none of them had evidence of MM or B-cell lymphoproliferative disorder.
In a retrospective study done by Nasr et al in 2009, entitled “Proliferative glomerulonephritis with monoclonal IgG deposits”, 37 patients were studied, of which 60% were older than 50 years. This study represented one of the largest series of PGNMID [2].
On SPEP and UPEP with immunofixation electrophoresis (IFE), seven patients had a monoclonal spike (M-spike) in both serum and urine, and four patients had an M-spike detectable in the serum only. The M-spike was detected at presentation in 10 of these 11 patients but not until repeat testing 3 years after presentation in the remaining patient. In all of these 11 patients, the monoclonal proteins in both serum and urine had the same heavy- and light-chain isotypes as the monoclonal Ig deposited in the kidney. IgG heavy-chain subclass analysis of glomerular deposits, performed in nine of the 11 patients, showed IgG1 in five patients, IgG2 in two, and IgG3 in two. Of the remaining 26 patients who had negative SPEP, UPEP, and IFE, four underwent serum free light-chain assay; of these, three were found to have normal κ:λ ratio, and one (who had glomerular monoclonal IgG3κ deposition) had an elevated κ:λ ratio. In our case, the patient had negative SPEP, UPEP, and IFE and serum free light-chain assay revealed normal kappa/lambda ratio.
At presentation, all patients had proteinuria. The mean 24-h urine protein was 5.70 g (range 0.36 - 17.00 g). Proteinuria was in the nephrotic range in 68.6% of patients, and 48.6% developed full nephrotic syndrome. Microhematuria was documented in 77.1% of patients. In our case, the patient had a urine protein/creatinine ratio of 0.36 mg/mg and had microhematuria persistent on repeated testing.
Regarding pathologic findings, the most common histologic pattern seen on LM was predominantly membranoproliferative GN characterized by diffuse and global double-contoured glomerular capillary walls (GCWs) with mesangial cell interposition and mesangial expansion by increased mesangial cell number and matrix. Most of these cases also showed endocapillary hypercellularity, including focal macrophage infiltration. The second most common pattern was predominantly endocapillary proliferative GN, characterized by endocapillary hypercellularity and leukocyte infiltration. IF identified staining of GBM for subclasses of IgG (IgG3-kappa being the most frequent type). In all cases, granular electron-dense deposits were confined to the glomerular compartment. The glomerular immune deposits were primarily subendothelial and mesangial. Subepithelial deposits were less frequent, and were segmental in most cases. In our case, LM showed an acute proliferative and exudative GN and EM showed glomeruli with endocapillary proliferation and neutrophilic proliferation and revealed subepithelial hump-shaped immune-type electron dense deposits (EDD) as well as mesangial and subendothelial deposits. IF revealed monoclonal IgG3-kappa deposits.
In our case, the findings of acute proliferative and exudative GN (Figs. 1 and 2) and the subepithelial hump-shaped (Fig. 3) immune-type electron dense deposits on LM and EM, respectively, would be most characteristic of acute post-infectious glomerulonephritis (APIGN). Interestingly, the lack of hypocomplementemia would be somewhat unusual for APIGN, and the immunofluorescence findings of IgG kappa restricted deposits are also atypical. The presence of deposits of a single Ig class which is IgG and a single light-chain, kappa, and the absence of other Igs, make the diagnosis of PGNMID most likely in our case (Fig. 4).
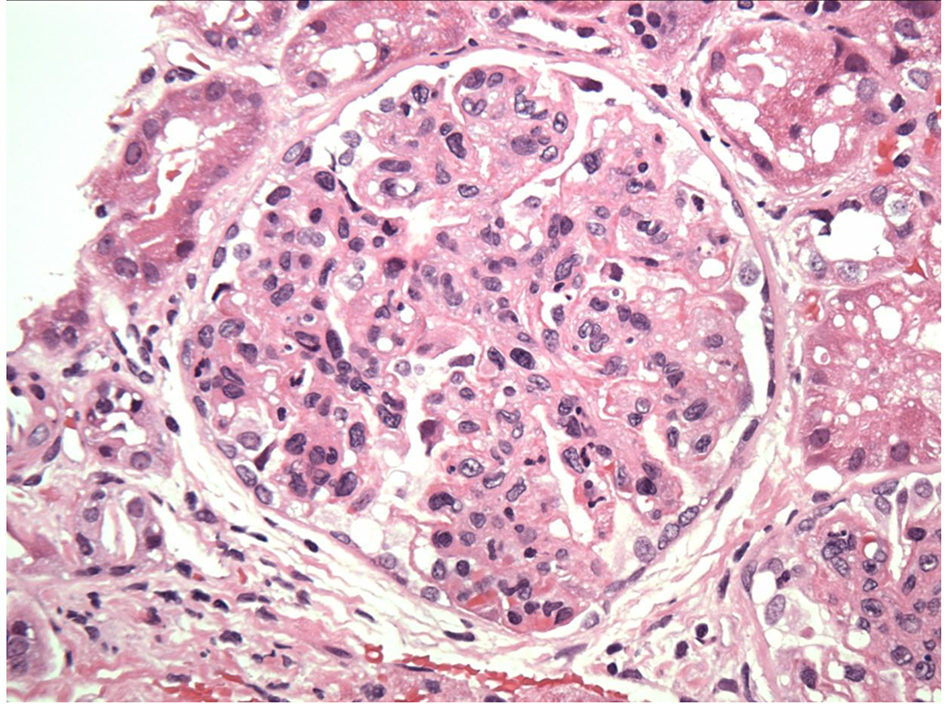 Click for large image | Figure 1. Exudative GN on light microscopy. |
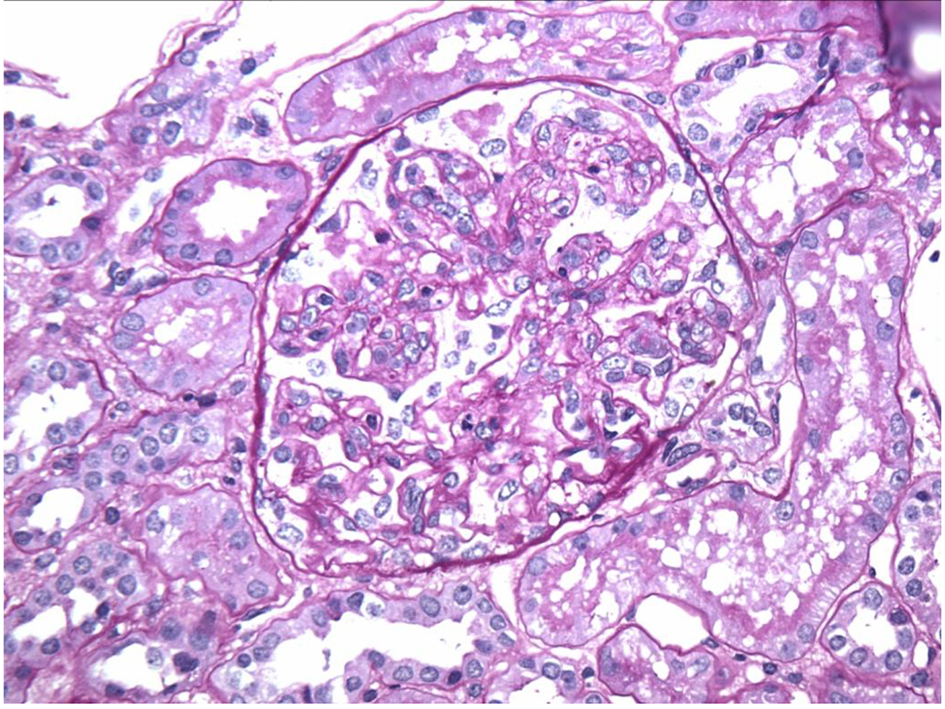 Click for large image | Figure 2. Endocapillary proliferation on light microscopy. |
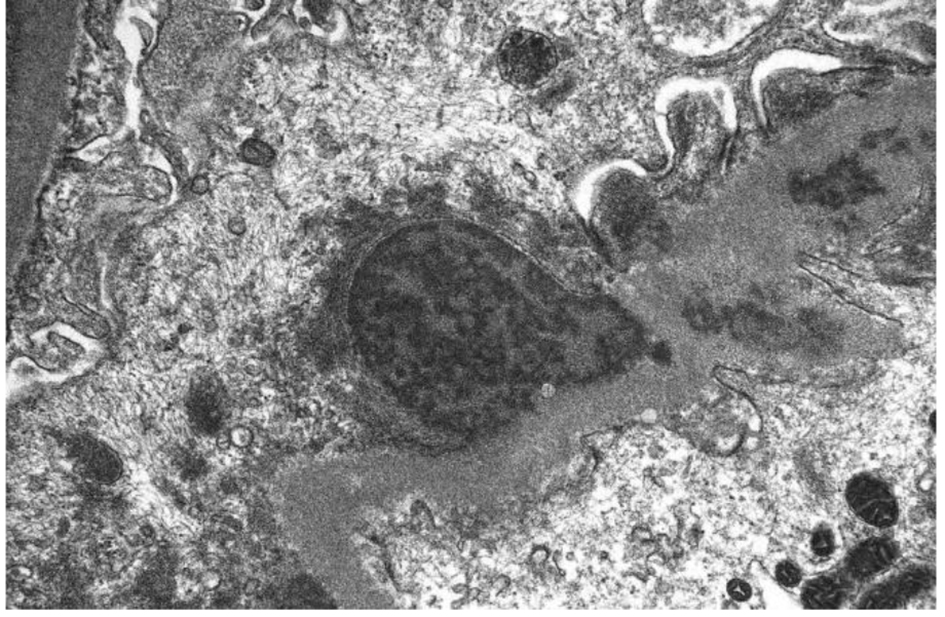 Click for large image | Figure 3. Subepithelial hump-shaped immune-type electron dense deposits on EM. |
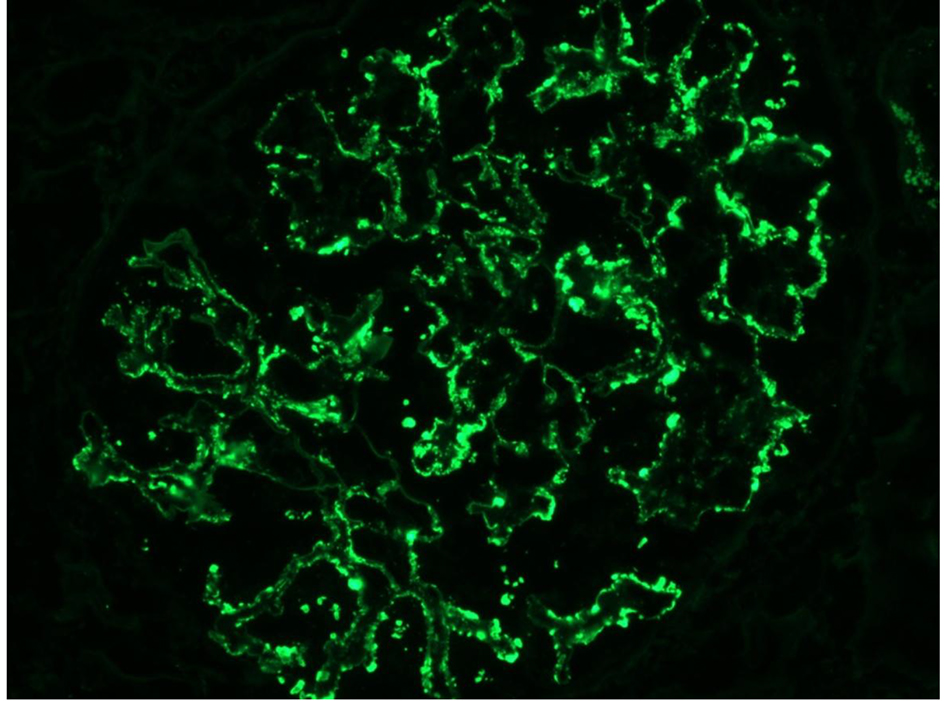 Click for large image | Figure 4. Monoclonal IgG kappa deposits on IF. |
In fact, the diagnosis of PGNMID is based on the following criteria [2]: findings on kidney biopsy of GN with 1) glomerular immune deposits staining positive for heavy-chain IgG, with negativity for Inga and IgM heavy-chains, thus confined to a single Ig class; 2) positive staining for a single IgG subclass (IgG1, IgG2, IgG3, or IgG4); 3) positive staining for a single light-chain isotype indicating monoclonality; 4) predominantly granular EDD in mesangial, subendothelial and/or subepithelial locations by EM, resembling immune complex GN; and 5) no clinical or laboratory evidence of cryoglobulines.
The etiology of PGNMID is not fully established. The majority of patients do not appear to have or develop hematologic malignancies during the published follow-up period. A small percentage of PGNMID cases were associated with neoplasm or viral infections [3-6]. In fact, there are 21 more patients with PGNMID that have been reported in the literature [7-9]. One patient reported by Evans et al [8] with PGNMID with monoclonal IgG-κ deposits was found to have a follicular B-cell lymphoma infiltrating the kidney and expressing surface IgG-κ. That patient had a positive Bence Jones protein in the urine as well as a monoclonal IgM protein on serum IFE. None of the remaining 20 patients had any hematologic malignancy. In our case, the patient had no detectable M protein by SPEP/UPEP/IFE.
In regards to the clinical outcome of patients with PGNMID, prognosis is variable. For instance, most of the 16 patients with available follow-up data were treated with prednisone with or without other immunosuppressive agents. Five out of the 16 patients subsequently progressed to ESRD (31.3%). In the study by Nasr et al [2], on an average of 30 months of follow-up for 32 patients, 38% had complete or partial recovery, 38% had persistent renal dysfunction, and 22% progressed to ESRD. Correlates of ESRD were higher creatinine, percentage of glomerulosclerosis, and degree of interstitial fibrosis but not immunomodulatory treatment or presence of a monoclonal spike. In our case, the patient had a complete remission with prednisone therapy.
Conclusion
In summary, we describe a unique case of PGNMID occurring in a young patient following a viral illness with spontaneous remission. The findings of an acute proliferative and exudative GN and the subepithelial hump-shaped immune-type electron dense deposits in the absence of hypocomplementemia, along with the presence of IgG kappa deposits, make the diagnosis of PGNMID most likely in our case. The pathophysiology of this disease is not well understood. Is PGNMID another form of post-infectious glomerulopathy in which an infection activates or induces a transient/acute monoclonal Ig formation?
| References | ▴Top |
- Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, Aucouturier P, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65(1):85-96.
doi pubmed - Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, Stokes MB, Nadasdy T, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20(9):2055-2064.
doi pubmed - Guiard E, Karras A, Plaisier E, Duong Van Huyen JP, Fakhouri F, Rougier JP, Noel LH, et al. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol. 2011;6(7):1609-1616.
doi pubmed - Barbour SJ, Beaulieu MC, Zalunardo NY, Magil AB. Proliferative glomerulonephritis with monoclonal IgG deposits secondary to chronic lymphocytic leukemia. Report of two cases. Nephrol Dial Transplant. 2011;26(8):2712-2714.
doi pubmed - Fujita E, Shimizu A, Kaneko T, Masuda Y, Ishihara C, Mii A, Higo S, et al. Proliferative glomerulonephritis with monoclonal immunoglobulin G3kappa deposits in association with parvovirus B19 infection. Hum Pathol. 2012;43(12):2326-2333.
doi pubmed - Yamada T, Arakawa Y, Mii A, Kashiwagi T, Kaneko T, Utsumi K, Masuda Y, et al. A case of monoclonal immunoglobulin G1-lambda deposition associated with membranous feature in a patient with hepatitis C viral infection. Clin Exp Nephrol. 2012;16(3):468-472.
doi pubmed - Alpers CE, Tu WH, Hopper J, Jr., Biava CG. Single light chain subclass (kappa chain) immunoglobulin deposition in glomerulonephritis. Hum Pathol. 1985;16(3):294-304.
doi - Evans DJ, Macanovic M, Dunn MJ, Pusey CD. Membranous glomerulonephritis associated with follicular B-cell lymphoma and subepithelial deposition of IgG1-kappa paraprotein. Nephron Clin Pract. 2003;93(3):c112-118.
doi pubmed - Komatsuda A, Masai R, Ohtani H, Togashi M, Maki N, Sawada K, Wakui H. Monoclonal immunoglobulin deposition disease associated with membranous features. Nephrol Dial Transplant. 2008;23(12):3888-3894.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.