

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 6, Number 8, August 2015, pages 362-366
Oligomeganephronia in an Adult Presenting With Features of Chronic Kidney Disease
Yoichi Iwafuchia, f, Takashi Moritab, Tetsuo Moriokac, Takeshi Kamurad, Yuko Oyamaa, Ichiei Naritae
aDepartment of Internal Medicine, Koseiren Sanjo General Hospital, 5-1-62 Tsukanome, Sanjo 955-0055, Japan
bDepartment of Pathology, Shinrakuen Hospital, 3-3-11 Shindoriminami Nishi-ku, Niigata 950-2087, Japan
cDepartment of Internal Medicine, Kidney Center, Shinrakuen Hospital, 3-3-11 Shindoriminami Nishi-ku, Niigata 950-2087, Japan
dDepartment of Radiology, Shinrakuen Hospital, 3-3-11 Shindoriminami Nishi-ku, Niigata 950-2087, Japan
eClinical Nephrology and Rheumatology, Niigata University Graduate School of Medical and Dental Sciences, 1-754 Asahimachi-Dori, Chuo-ku, Niigata 951-8510, Japan
fCorresponding Author: Yoichi Iwafuchi, Department of Internal Medicine, Koseiren Sanjo General Hospital, Tsukanome 5-1-62, Sanjo 955-0055, Japan
Manuscript accepted for publication June 30, 2015
Short title: Oligomeganephronia in an Adult
doi: http://dx.doi.org/10.14740/jmc2212w
| Abstract | ▴Top |
We report the case of a 29-year-old man with oligomeganephronia (OMN) who presented with symptoms of chronic kidney disease (CKD). Radiological and ultrasound images showed bilateral mildly small and uneven kidneys. Right renal biopsy was performed. Under light microscopy, the number of glomeruli in histological samples was decreased and the remaining glomeruli were markedly enlarged without mesangial proliferation. The obsolescent glomeruli had findings compatible with segmental sclerosis. Tubules were dilated and enlarged, and there was a presumptive primitive duct enclosed by a smooth muscle collar. Dysplastic changes in the biopsy specimen suggested that the cause of the patient’s small kidneys was a congenital anomaly rather than acquired kidney disease. OMN is one of the probable causes of CKD in adults. Angiotensin type II receptor blockers may be an effective treatment for focal segmental glomerulosclerosis due to OMN.
Keywords: Oligomeganephronia; Adult case; Chronic kidney disease; Focal segmental glomerulosclerosis; Renal hypodysplasia; Primitive duct; Angiotensin type II receptor blocker
| Introduction | ▴Top |
Oligomeganephronia (OMN) is a congenital non-familial anomaly of bilateral renal hypoplasia, first described by Habib et al in 1962 and characterized by a striking reduction in the number of nephrons and markedly enlarged glomeruli [1]. Children with OMN develop polyuria, polydipsia, urine concentrating deficiency, and proteinuria, usually leading to progressive renal failure during childhood [2]. Some cases are initially considered asymptomatic proteinuria, with diagnosis of OMN based on histologic examination occurring by school age. We report the diagnosis of OMN in an adult man presenting with symptoms of chronic kidney disease (CKD), who had marked proteinuria that improved with the administration of an angiotensin type II receptor blocker (ARB).
| Case Report | ▴Top |
A 29-year-old man was referred to our hospital because of a 12-year history of proteinuria. In the 5 years before admission, his renal function had gradually worsened. The patient was admitted to our hospital for diagnostic evaluation and treatment. He had been born full term, with a normal birth weight of 3,100 g. He was diagnosed with a ventricular septal defect (VSD) at 5 years of age. The patient’s growth and development had been otherwise normal. He had no history of recurrent urinary tract infections. There was no family history of renal disease.
Physical exam findings were as follows: height 178.0 cm, weight 80.6 kg, body mass index 25.4, blood pressure 126/68 mm Hg, pulse rate 72 beats/min, and body temperature 36.6 °C. The patient was not deaf. The patient’s ocular fundus was normal. The remainder of his physical examination was unremarkable, without ascites or pretibial edema.
Urinary protein was 1,105 mg/dL and 3.8 g/day. The protein/creatinine ratio in a spot urine specimen was 3.4. The urinary sediment showed 1 - 4 erythrocytes and 1 - 4 leukocytes per high-power field. The patient’s hematocrit was 45.6%, his hemoglobin concentration was 15.5 g/dL, his platelet count was 205,000/µL, and his leukocyte count was 5,590/µL. His serum urea nitrogen level was 15.2 mg/dL, creatinine was 1.61 mg/dL, estimated glomerular filtration rate (eGFR) was 43.8 mL/min/1.73 m2, uric acid was 9.8 mg/dL, cholesterol was 172 mg/dL, total protein was 6.7 g/dL, and albumin was 3.8 g/dL. His C-reactive protein level was 0.05 mg/dL, IgG was 918 mg/dL, IgA was 138 mg/dL, IgM was 74 mg/dL, and ferritin was 129 ng/mL. Serum complement was normal. Circulating immune complexes were negative. The other autoimmune serological findings were within normal ranges and serum hepatitis viral markers were negative. An intravenous pyelogram revealed smaller (right) and small (left) kidneys without hydronephrosis. Size of renal pelvis and calices of bilateral kidneys seemed mildly small (Fig. 1). Abdominal computed tomography (Fig. 1) and ultrasound showed smaller (right) and small (left) kidneys with uneven surface of kidneys (longitudinal diameters: right kidney 10.1 cm and left kidney 8.8 cm on ultrasonography), but apparent thinning of bilateral renal cortex was not present. Electrocardiography revealed normal sinus rhythm with normal axis and intervals. Echocardiography revealed a small VSD with left to right shunting.
 Click for large image | Figure 1. An intravenous pyelogram had revealed smaller (right) and small (left) kidneys without hydronephrosis. Size of renal pelvis and calices of bilateral kidneys seemed mildly small. Abdominal computed tomography showed uneven surface of smaller (right) and small (left) kidneys without apparent thinning of renal cortex. |
On the patient’s second day in the hospital, a right renal biopsy was performed to investigate the cause of proteinuria. Under light microscopy, the number of glomeruli in the biopsy sample was obviously decreased, with only nine glomeruli found in two pieces of adequate length. Of these glomeruli, one was obsolescent, one was becoming obsolescent, and the remaining glomeruli were markedly enlarged with slightly hyperplastic juxtaglomerular apparati, but without mesangial proliferation (Fig. 2a). Diameters of two glomeruli were 300 μm and 320 μm. The glomerulus in the process of becoming obsolescent had findings compatible with segmental sclerosis; segmental sclerotic lesions, adhesion of glomerular tufts, crescent-like epithelial cell proliferation, and hyaline deposits were seen (Fig. 2b). Tubules were dilated and enlarged, and focal tubular atrophy with mild interstitial cellular infiltration was noted (Fig. 2c). There was a peculiar area in the cortex containing numerous small vessels, scattered muscle-like cells, and a presumptive primitive duct enclosed by a smooth muscle collar (Fig. 2c, d). The epithelial cells of this duct were cytokeratin positive (Fig. 2e) and the collar cells were smooth muscle actin-positive (Fig. 2f). No significant deposits of immunoglobulins or complement components were found with immunofluorescence. Only one glomerulus was found in the electron microscopic specimen and neither electron dense deposits nor specific findings were observed. These imaging and histological findings led us to the diagnosis of OMN.
 Click for large image | Figure 2. (a) An enlarged glomerulus with slightly hyperplastic juxtaglomerular apparatus but without proliferative changes or thickening of the glomerular basement membrane (periodic acid-methenamine silver, original magnification × 28). (b) An obsolescent glomerulus with thickened Bowman’s capsule, adhesion of tufts, crescent-like proliferation of epithelial cells, hyaline deposits (H), and remaining open tufts (periodic acid-methenamine silver, original magnification × 68). (c) Some tubules are atrophic with a thickened tubular basement membrane, while other tubules are relatively enlarged. In the center, a peculiar area contains a presumptive primitive duct (P) and numerous small vessels (periodic acid-methenamine silver, original magnification × 20). (d) A presumptive primitive duct enclosed by a smooth muscle collar (hematoxylin-eosin, original magnification × 80). (e) Epithelial cells of the presumptive primitive duct (P) are cytokeratin positive, as are other tubular epithelial cells (immunostain with Becton Dickinson-CAM5.2, original magnification × 40). (f) The muscle collar of the primitive duct (P) and the walls of small vessels and scattered muscle-like cells are positive for smooth muscle actin (Immunostain with DAKO-anti-smooth muscle actin antibody, original magnification × 22). |
After obtaining informed consent, we collected DNA from the patient. Using his whole blood, we looked for mutations in paired box gene 2 (PAX2) and hepatocyte nuclear factor-1 beta (HNF-1β), but no mutations were detected.
The patient was treated with 20 mg/day of valsartan in our outpatient clinics. After administration of valsartan, his proteinuria appreciably reduced. Two years later, his urine protein/creatinine ratio had reduced to 1 - 1.4 and his eGFR had stayed at 52.1 mL/min/1.73 m2.
| Discussion | ▴Top |
Renal biopsy-proven cases of OMN are very rare in adults. We have identified only six reported adult cases [3-5] confirmed by renal biopsy, including the case presented here (Table 1). These six cases included five males and one female with a mean age of 28 years (range 19 - 36 years). Low birth weight and intrauterine growth restriction are sometimes related to OMN, but five of the six patients were born at full term with normal birth weights. Two patients had hypertension, three had proteinuria of less than 1.0 g/day, and three had proteinuria over 3.0 g/day. The median serum creatinine level at presentation was 1.71 mg/dL, and the median eGFR was 44.3 mL/min/1.73 m2. The present case was an adult with OMN, who showed marked proteinuria, moderate renal failure, and bilateral atrophic and uneven kidneys.
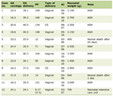 Click to view | Table 1. Reported Cases of Oligomeganephronia in Adults |
The pathogenesis of OMN remains unknown, but OMN has been observed in association with certain genetic disorders, including mutations in the PAX2 [6] and HNF-1β [7] genes. OMN associated with a PAX2 mutation has been reported as one of the symptoms of renal-coloboma syndrome [8]. Mutation of the PAX2 gene associated with OMN without coloboma has also been reported [9]. We did not detect mutations of PAX2 or HNF-1β in our patient.
OMN is usually a sporadic isolated malformation, but it has sometimes been reported with other congenital malformations, such as urinary tract anomalies and deformities of the hands, feet, or ears [6, 10]. One of the reported adult cases had complete hearing loss in the left ear (Table 1). In addition to clinical and histological findings, concurrent congenital malformations are helpful in arriving at a diagnosis of OMN. In our patient, a small VSD was observed. This is the first reported case of OMN with concurrent VSD. It is thought that OMN results from developmental arrest of the metanephric renal blastoma between the 14th and 20th weeks of fetal life [11]. Although it is possible that common factors influenced the development of renal and cardiac abnormalities in our patient, further evaluation of a greater number of cases is needed.
The characteristic histological findings in OMN include decreased numbers of nephrons with hypertrophic glomeruli, tubules, and juxtaglomerular apparati [12, 13]. McGraw et al concluded that increasing proteinuria in patients with OMN heralds the development of focal segmental glomerulosclerosis (FSGS), presumably due to functional overload of the reduced nephron number [14]. These characteristic findings were observed in our patient.
OMN is generally classified as renal hypoplasia. Hypoplastic kidneys are defined as small kidneys weighing less than 50% of the normal mean for the patient’s age, and with no histological abnormality other than secondary FSGS, simple hypoplasia, or hypertrophy of nephrons (oligomeganephronic hyperplasia). In the case of our patient, a presumptive primitive duct enclosed by a smooth muscle collar, which is a distinct feature of renal dysplasia, was observed. To our knowledge, this is the first case of OMN with renal dysplasia, confirmed by needle biopsy. Comparing a figure of apparent infant case in a text book [15], cells enclosing primitive duct of our case appear more matured muscle cells, the possibility that mesenchymal cells in dysplasia can mature with age of patients will be determined by accumulation of such cases. The degree of hypoplasia in our patient was relatively mild, as in the five previously reported adult cases, and glomerular size was not large in comparison with typical juvenile OMN. In consequence, our patient’s renal dysfunction did not become apparent until adulthood. Our case showed relatively small kidneys with small renal pelvis and calices, but apparent thinning of bilateral renal cortex was not present.
The radiological findings of abdominal computed tomography in our patient looked like renal scar or reflux nephropathy. But existence of reflux nephropathy was obscure from his past history. The existence of dysplastic changes suggests that the cause of our patient’s small kidneys was a congenital anomaly rather than acquired kidney disease. As recently pointed out, progressive scarring of the kidneys has been observed in the absence of urinary tract infections and obstruction, and this probably represents primary renal dysplasia [16].
CKD is a worldwide public health problem, with adverse outcomes of kidney failure, cardiovascular disease, and premature death. In 2002, a simple definition of CKD was proposed and subsequently adopted worldwide. Our case had CKD stage G3bA3. Our patient’s proteinuria had continued for at least 12 years, and he had experienced none of the characteristic symptoms associated with OMN, such as polyuria, polydipsia, or growth retardation. The clinical course of our case matched the definition of CKD. OMN is one of the probable cause of CKD in adults, especially CKD with bilateral small and uneven kidneys. Patients with OMN sometimes have atrophic kidneys, which is a relative contraindication for renal biopsy. The definitive diagnosis of OMN must be based on renal histopathological findings, so OMN in adults may be overlooked. In cases without histological findings, it is difficult to distinguish between congenital and acquired disease.
Our patient’s proteinuria was appreciably reduced by ARB administration. In patients with glomerulomegaly and FSGS, angiotensin inhibitors, such as angiotensin converting enzyme inhibitors and ARB, decrease proteinuria and slow the rate of progression to end stage renal disease [17]. ARB might be effective in reducing proteinuria in FSGS due to OMN [4].
Acknowledgement
The authors are grateful to Dr. N. Morisada and Dr. K. Iijima (Department of Pediatrics, Kobe University Graduate School of Medicine) for their help with genetic analysis, and to Mr. N. Sakamoto, Ms. S. Tsuchida, Ms. M. Yoshinuma, and Ms. M. Igashima (Department of Pathology, Shinrakuen Hospital) for their technical assistance.
Conflict of interest
All of the authors have no competing interests to declare.
| References | ▴Top |
- Habib R, Courtecuisse V, Mathieu H, Royer P. [A peculiar anatomo-clinical type of chronic renal insufficiency in the child: bilateral congenital oligonephronic hypoplasia]. J Urol Nephrol (Paris). 1962;68:139-143.
- Drukker A. Oligonephropathy: from a rare childhood disorder to a possible health problem in the adult. Isr Med Assoc J. 2002;4(3):191-195.
pubmed - Kawanishi K, Takei T, Kojima C, Moriyama T, Sugiura H, Itabashi M, Tsukada M, et al. Three cases of late-onset oligomeganephronia. NDT Plus. 2011;4(1):14-16.
doi - Fuke Y, Hemmi S, Kajiwara M, Yabuki M, Fujita T, Soma M. Oligomeganephronia in an adult without end stage renal failure. Clin Exp Nephrol. 2012;16(2):325-328.
doi pubmed - Alves RJ, Oppermann K, Schein LE, Pegas KL. A case of late-onset oligomeganephronia. J Bras Nefrol. 2012;34(4):392-394.
doi pubmed - Salomon R, Tellier AL, Attie-Bitach T, Amiel J, Vekemans M, Lyonnet S, Dureau P, et al. PAX2 mutations in oligomeganephronia. Kidney Int. 2001;59(2):457-462.
doi pubmed - Sagen JV, Bostad L, Njolstad PR, Sovik O. Enlarged nephrons and severe nondiabetic nephropathy in hepatocyte nuclear factor-1beta (HNF-1beta) mutation carriers. Kidney Int. 2003;64(3):793-800.
doi pubmed - Schimmenti LA. Renal coloboma syndrome. Eur J Hum Genet. 2011;19(12):1207-1212.
doi pubmed - Nishimoto K, Iijima K, Shirakawa T, Kitagawa K, Satomura K, Nakamura H, Yoshikawa N. PAX2 gene mutation in a family with isolated renal hypoplasia. J Am Soc Nephrol. 2001;12(8):1769-1772.
pubmed - Yui I, Awazu M, Takeuchi Y, Fukuda J, Hanada T, Sakaguchi H. [Oligomeganephronia with multiple anomalies]. Nihon Jinzo Gakkai Shi. 1984;26(11):1529-1537.
pubmed - Woolf AS, Price KL, Scambler PJ, Winyard PJ. Evolving concepts in human renal dysplasia. J Am Soc Nephrol. 2004;15(4):998-1007.
doi pubmed - Fetterman GH, Habib R. Congenital bilateral oligonephronic renal hypoplasia with hypertrophy of nephrons [oligomeganephronie]. Studies by microdissection. Am J Clin Pathol. 1969;52:199-207.
- Morita T, Wenzl J, McCoy J, Porch J, Kimmelstiel P. Bilateral renal hypoplasia with oligomeganephronia: quantitative and electron microsopic study. Am J Clin Pathol. 1973;59(1):104-112.
pubmed - McGraw M, Poucell S, Sweet J, Baumal R. The significance of focal segmental glomerulosclerosis in oligomeganephronia. Int J Pediatr Nephrol. 1984;5(2):67-72.
pubmed - Heptinstall's Pathology of the Kidney. 6th ed. Philadelphia: Lippincott, 2007; p1294, Figure 26.50.
- Connolly JO, Neild GH. Congenital anomalies of the kidney and urinary tract. In: Johnson RJ, Freehally J, Floege J, editor. Comprehensive Clinical Nephrology. Philadelphia: Elisevier Saunder, 2015;613-630.
- Usta M, Ersoy A, Dilek K, Ozdemir B, Yavuz M, Gullulu M, Yurtkuran M. Efficacy of losartan in patients with primary focal segmental glomerulosclerosis resistant to immunosuppressive treatment. J Intern Med. 2003;253(3):329-334.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.