

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 6, Number 6, June 2015, pages 257-263
Perioperative Management of a Patient With Glutaric Aciduria
Mumin Hakima, David P. Martina, b, Allan Beebec, Jan Klamarc, Joseph D. Tobiasa, b, d
aDepartment of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, Columbus, OH, USA
bDepartment of Anesthesiology, The Ohio State University, Columbus, OH, USA
cDepartment of Orthopedic Surgery, Nationwide Children’s Hospital, Columbus, OH, USA
dCorresponding Author: Joseph D. Tobias, Department of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, 700 Children’s Drive, Columbus, OH 43205, USA
Manuscript accepted for publication April 29, 2015
Short title: Management of Glutaric Aciduria
doi: http://dx.doi.org/10.14740/jmc2096w
| Abstract | ▴Top |
Glutaric aciduria type-1 (GA-1) is an autosomal recessive metabolic disorder due to the deficiency of the enzyme, glutaryl-CoA dehydrogenase. The enzymatic defect leads to secondary damage to the central nervous system due to the accumulation of glutaric acid. Due to the progressive neurologic effects with spasticity and orthopedic deformities, surgical and anesthetic cares are frequently required. We present a 13-year-old girl with glutaric acidemia type 1 who required anesthetic care for posterior spinal fusion. Previous reports of anesthetic care for these patients are reviewed, the end-organ involvement is discussed, and options for anesthetic care are presented.
Keywords: Posterior spinal fusion; Scoliosis; Glutaric aciduria type-1
| Introduction | ▴Top |
Glutaric aciduria type-1 (GA-1) is an uncommon, severe autosomal recessive metabolic disorder due to the deficiency of the enzyme, glutaryl-CoA dehydrogenase (GCDH) within the mitochondria [1]. It is one of several genetic/metabolic disorders in the category of organic acidurias. The disorder generally presents in early childhood with the majority of children having no measurable enzyme activity. The estimated prevalence varies from 1 in 30,000 newborns in one Scandinavian study to 1 in 30,000 - 100,000 newborns in other studies [2-5]. The prevalence may be much higher in isolated populations in the Middle East countries due to higher rates of consanguinity [6, 7]. GA-1 was described first by Goodman et al in 1975 [8]. Mutations in the mitochondrial GCDH gene located on chromosome 19 (19p13.2) lead to a deficiency of GCDH, a mitochondrial enzyme involved in the metabolism of lysine, hydroxyl-lysine, and tryptophan [8-11]. This defect results in the accumulation of GA and 3-hydroxyglutaric acid (3-OHGA) with secondary carnitine depletion. The progressive neurologic effects of the disorder frequently lead to spasticity and orthopedic deformities requiring surgical and anesthetic care. We present a 13-year-old girl with glutaric acidemia type 1 who required anesthetic care for posterior spinal fusion. Previous reports of anesthetic care for these patients are reviewed, the end-organ involvement is discussed, and options for anesthetic care are presented.
| Case Report | ▴Top |
Institutional Review Board approval is not required at Nationwide Children’s Hospital (Columbus, OH) for the presentation of single case report. The patient was a 13-year-old, 31.75 kg, girl who presented for posterior spinal fusion for the treatment of progressive scoliosis. Her past history was significant for GA-1 which was diagnosed at 5 months of age. Her past surgical history included Nissen fundoplication with gastrostomy tube (G-tube) placement due to aspiration problems, umbilical hernia repair, bilateral tympanostomy with tube placement, left hip joint surgery, and botulinum toxin injections to treat spasticity. Associated comorbid conditions included gastroesophageal reflux, dysphagia with chronic vomiting, osteoporosis, central nervous system (CNS) involvement with spasticity, osteoporosis, developmental delay, asthma, bowel and urinary incontinence. Current home medications included levocarnitine (500 mg via G-tube once daily), riboflavin (100 mg via G-tube every night), clonazepam (0.5 mg via G-tube TID), diazepam (5 mg via G-tube), diazepam rectal gel as needed for seizures lasting more than 5 min, lamotrigine (75 mg in the morning and 100 mg at night), lansoprazole (30 mg once a day), sucralfate (500 mg via G-tube TID), ondansetron (3 mg via G-tube QID), albuterol (high-flow nebulizer every 4 h), and ergocalciferol (1,000 units via G-tube once a day). Allergies included oxcarbazepine. Preoperative physical examination revealed an adolescent in no acute distress with moderate scoliosis, normal systemic examination, and a G-tube in place. Airway examination revealed a Mallampati grade I view. Preoperative laboratory evaluations including electrolytes, renal function, coagulation function, blood glucose and hepatic function were normal. The hemoglobin was 10.2 g/dL with a hematocrit of 32.2%. The patient was admitted the day before surgery to manage her preoperative comorbid conditions. She was held nil per os (NPO) for 6 h except for her usual medications and a peripheral intravenous infusion of 5% dextrose in 1/2 normal saline was started at 1.5 times maintenance. The preoperative blood glucose was 79 mg/mL with a room air oxygen saturation of 99%. The patient was transported to the operating room and routine American Society of Anesthesiologists’ monitors were applied. Anesthesia was induced with propofol (50 mg) and fentanyl (25 µg) which were administered intravenously through a pre-existing peripheral intravenous line. Bag-valve-mask ventilation was provided without difficulty. Neuromuscular blockade was provided with rocuronium (30 mg) and direct laryngoscopy was performed with a Miller 2 blade. Laryngoscopy revealed a Cormack-Lehane grade I view and a 6.0 mm cuffed endotracheal tube was placed on the first attempt. After the induction of anesthesia, arterial and central venous accesses were obtained using ultrasound guidance. Additionally, using ultrasound guidance two additional 18-gauge intravenous cannulas were placed in the right and left saphenous veins. Tranexamic acid was administered for prevention of fibrinolysis and limit intraoperative blood loss (50 mg/kg followed by an infusion at 5 mg/kg/h). The patient was turned and positioned prone. Baseline neurophysiological monitoring including motor evoked potentials (MEP) and somatosensory evoked potentials (SSEPs) was obtained. Per our usual practice to allow for neurophysiological monitoring during spinal surgery, anesthesia was maintained with desflurane titrated to maintain the bispectral index (BIS) at 50 - 60 and a remifentanil infusion of 0.1 - 0.3 µg/kg/min to maintain the mean arterial pressure at 55 - 65 mm Hg [12]. Intraoperative antibiotics included cefazolin and gentamicin. The MAP was maintained at 55 - 65 mm Hg for controlled hypotension to minimize intraoperative blood loss. Heart rates varied from 60 to 120 beats per minute with a normal sinus rhythm. No bradycardia or arrhythmias were noted. During the procedure, the amplitude of the MEP was noted to decrease during placement of spinal instrumentation. A phenylephrine infusion was started to maintain the MAP ≥ 70 mm Hg, which resulted in the return of the MEP to baseline. Intraoperative fluids included 1 unit of packed red blood cells, 125 mL of cell saver autologous blood, 250 mL of 5% albumin and 2,007 mL of isotonic crystalloid solution. The estimated blood loss was 600 mL. During wound closure, the remifentanil and tranexamic acid infusions were discontinued. Acetaminophen (15 mg/kg) was administered intravenously along with incremental doses of hydromorphone to provide postoperative analgesia. Following completion of the surgical procedure, the patient was turned supine and her trachea was extubated when awake. The patient was transferred to the post-anesthesia care unit (PACU) followed by the pediatric intensive care unit (PICU) for observation of hemodynamic and respiratory function. Postoperative pain control was provided with hydromorphone delivered via nurse-controlled analgesia. The remainder of her postoperative course was uncomplicated and she was discharged home on postoperative day 5.
| Discussion | ▴Top |
GA-1 is an inherited metabolic disorder resulting from a deficiency of the mitochondrial enzyme, GCDH. This enzyme is involved in the degradation of several amino acids (lysine, hydroxyl-lysine, and tryptophan) with their eventual conversion to acetyl-CoA and entry into the Kreb’s cycle as citric acid. The absence of this enzyme results in increased levels of glutaric acid and a related metabolite (quinolinic acid) with reduced levels of γ-amino-butyric acid (GABA) within the brain and cerebrospinal fluid. These alterations result in progressive damage to the CNS, especially in the basal ganglia (caudate nucleus and putamen) resulting in extrapyramidal symptoms and seizures. Routine newborn screening has allowed early diagnosis with the institution of dietary restriction, carnitine and riboflavin supplementation, and treatment of acute encephalopathic crisis, thereby allowing for more normal development [11, 13, 14]. Despite these therapies, acute exacerbations of the disorder (encephalopathic crisis) may be triggered by infections, stress, fever, or surgery. These crises may result in progressive CNS damage.
Depending on the age of diagnosis, there can be a wide spectrum of clinical features in patients with GA-1 including severe and progressive CNS involvement with severe psychomotor retardation [15]. Macrocephaly is generally present at birth; however, progressive CNS damage can lead to microcephaly [16]. Without routine newborn screening, early diagnosis is difficult as there are no pathognomonic signs or symptoms. Without clinical treatment including dietary therapy, the majority of patients manifest clinical signs and symptoms by 6 - 12 months of age including psychomotor delay, dystonia, and seizures [17, 18]. Hypotonia may be present early, progressing to spastic quadriparesis with progressive CNS involvement. An encephalopathic crisis with necrosis of the basal ganglia can result in dystonic dyskinetic movement disorders such as dysarthria and choreoathetosis, which may be misdiagnosed as athetoid cerebral palsy, attributed to perioperative birth asphyxia. Although there are no specific radiologic findings, CNS imaging may reveal suggestive clinical features including hydrocephalus, brain atrophy, structural changes of basal ganglia, and demyelization. In the majority of patients, basal ganglia abnormalities, the incomplete opercularization of the insular cortex with widening of the Sylvian fissures and CSF spaces can be seen. Additionally, subdural collections of fluid over the convexities with bilateral temporal arachnoid cysts have been reported [19, 20]. Subdural hematomas suggestive of non-accidental trauma have been reported, making it mandatory to rule out GA-1 in cases of suspected non-accidental trauma [21, 22]. If not identified during routine newborn screening, the diagnosis can be confirmed by quantification of 3-OHGA in urine. However, some patients may not excrete large amounts of 3-OHGA and therefore, direct measurement of GCDH activity in fibroblasts and lymphocytes provides the most accurate diagnostic modality [23].
Given the significant comorbid sequelae of GA-1, there are several specific perioperative implications which may significantly impact the risk for perioperative morbidity and mortality. As with the anesthetic care of all patients, the focus of effective perioperative care begins with the preoperative examination and the identification of end-organ involvement by the primary disease process. Previous reports regarding the perioperative care of patients with GA-1 syndrome are summarized in Table 1 [24-29]. Perioperative care focuses on the maintenance of normal homeostasis with provision of fluid and glucose during fasting to prevent catabolism and the release of endogenous amino acids. During NPO times, given the potential impact of catabolism and the risks of hypoglycemia, intravenous fluids and dextrose should be provided to meet maintenance needs. Perioperatively, routine medications including anticonvulsant agents, carnitine, and riboflavin should be continued.
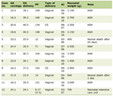 Click to view | Table 1. Previous Reports of Anesthetic Care for Patients With Glutaric Aciduria |
When considering the case reports listed in Table 1, they demonstrate several features which should be considered when anesthetizing patients with GA. Most chose to start a peripheral intravenous infusion of glucose to avoid catabolism when the children were placed NPO. The glucose infusion was continued intraoperatively and postoperatively in many patients with ongoing systemic glucose monitoring. In general, they noted no problems with airway management either by bag-valve-mask ventilation or direct laryngoscopy and endotracheal intubation. Several chose to use cricoid pressure or a modified rapid sequence intubation given the potential issues of gastroesophageal reflux in children with chronic debilitating CNS disorders. These case reports demonstrate the safe use of both volatile anesthetic agents and propofol. Intermediate acting neuromuscular blocking agents (NMBAs) such as cis-atracurium or rocuronium were used with no reports of the use of succinylcholine. In one report, brief CNS imaging was accomplished as an outpatient procedure with intravenous propofol sedation while the other patients were admitted at least overnight for monitoring.
One of the primary concerns in children with GA-1 or any chronic debilitating CNS disorder with loss of function relate to the possibility of pulmonary aspiration during anesthetic induction, prolonged responses to non-depolarizing NMBAs, and a hyperkalemic response to succinylcholine. Dysphagia is often seen in these cases, especially with severe dystonia which results in repeated chocking and regurgitation, increasing the risk for aspiration of gastric contents during general anesthesia. Precautions to prevent such problems may include the preoperative administration of H2-antagonists blockers and the use of cricoid pressure with modified rapid-sequence induction. Medications with extrapyramidal side effects such as metoclopramide and droperidol, although sometimes used to facilitate gastric emptying or treat nausea, should be avoided given their extrapyramidal effects in a patient with underlying involvement of the basal ganglia.
For airway management, a rapidly acting NMBA may be required. Although higher doses of rocuronium (1 mg/kg) may speed the onset and allow earlier tracheal intubation, an exaggerated and prolonged response may be seen in patients with CNS disorders with hypotonia. Given the associated involvement of the CNS, the use of succinylcholine is controversial because of possible hyperkalemia [30]. However, the associated hypotonia may lead to an exaggerated response to NMBAs, making precise dosing with train-of-four monitoring a prerequisite to avoid prolonged recovery times. Residual neuromuscular blockade should be reversed at the completion of the procedure and full recovery documented prior to tracheal extubation. Although not available in the United States, sugammadex may offer an advantage in patients with pre-existing hypotonia who require reversal from profound blockade.
Issues related to poor upper airway control and defective control of ventilation with the associated CNS damage may lead to perioperative respiratory insufficiency. Such problems may be exacerbated by the residual effects of anesthetic agents, the surgical procedure, poor cough effort, and the use of postoperative opioids for pain management. As noted in the previous reports of anesthetic care in patients with GA, short acting anesthetic agents (desflurane, sevoflurane, and remifentanil) may be chosen to eliminate concerns of their effects on postoperative respiratory function. In our patient, the choice of maintenance anesthetic agents including desflurane and remifentanil was based on not only the comorbid conditions, but also the intraoperative requirements mandated by the surgical procedure and neurophysiological monitoring [12]. Given the variable response to anesthetic agents and the need to maintain an acceptable level of anesthesia in a patient with CNS damage, we chose to use bispectral index monitoring to gauge the depth of anesthesia while using remifentanil to provide analgesia and control the hemodynamic response [31, 32]. Postoperative monitoring of respiratory function is suggested especially when opioids are required for postoperative analgesia. The use of adjunctive agents (acetaminophen or non-steroidal anti-inflammatory agents) is suggested as a means of decreasing opioid needs and their associated adverse effects. Non-invasive techniques of respiratory support such as BiPAP may be used to facilitate postoperative tracheal extubation in these patients and avoid postoperative respiratory insufficiency [33, 34].
Although there have been anecdotal reports of malignant hyperthermia in patients with enzymatic defects involving the mitochondria, the association is not universal and the use of volatile agents has been demonstrated to be safe and as demonstrated in the reports reviewed in Table 1 has been shown to be safe and effective in patients with GA [35-38]. Of note, carnitine deficiency is a frequent secondary finding in patients with GA. As carnitine is an essential cofactor in the transport of long-chain fatty acids into the mitochondria and has a pivotal role in fatty acid oxidation, it has been suggested that propofol should be avoided in this condition. Propofol not only can provide excessive lipid, in select patients, it also may impair mitochondrial electron transport with inhibition of oxidative phosphorylation, carnitine palmitoyltransferase transport of long-chain fatty acids, and β-oxidation of fatty acids in the mitochondria [39]. This could predispose to propofol infusion syndrome and severe metabolic acidosis in patients with mitochondrial disorders, carnitine deficiency and inadequate carbohydrate intake [40, 41]. Therefore, the use of propofol for prolonged anesthetic care remains controversial in patients with mitochondrial disorders. Although its use as a single dose for anesthetic induction has not been questioned, the safety of more prolonged infusions remains questionable with some authors cautioning against its uses. Given the potential for inhibition of mitochondrial function, it may be appropriate to monitor acid-base status and plasma lactic acid when prolonged infusions are used and to immediately discontinue the infusion should chemical abnormalities be noted [42].
Given the progressive deterioration of CNS function, seizures are a frequent comorbid condition in patients with GA-1. Preoperative management to limit the potential for perioperative seizures includes optimizing and confirming therapeutic anticonvulsant levels prior to the surgical procedure. Routine anticonvulsant medications should be administered the morning of the procedure despite concerns of the patient’s NPO status with subsequent intraoperative dosing as needed [43]. Alternative routes of delivery (intravenous or rectal) may be required when enteral administration is not feasible. Consultation with the neurology or pharmacology service may be helpful to determine dosing conversion from enteral to intravenous administration or to guide intraoperative redosing. Sodium valproate inhibits mitochondrial β-oxidation of fatty acids and should be avoided in patients with mitochondrial metabolic disorders [44, 45].
Although it has known that specific agents such as etomidate may activate the EEG and even stimulate seizure activity, the inhalational anesthetic agents, propofol, and the barbiturates have potent anticonvulsant properties. Many of these agents have been used to treat status epilepticus that is refractory to conventional therapy [46]. Although motor movements resembling seizure activity and even occasional spike and wave activity on the EEG have been reported with sevoflurane, these effects generally occur only when the inspired concentration is rapidly increased during anesthetic induction when there is accompanying hypocarbia [47, 48]. In general clinical practice, there does not appear to be any contraindication to the use of sevoflurane in this population and other patients with underlying seizure disorders.
Patients with progressive and deteriorating neurologic disorders such as GA-1 frequently have multiple joint contractures. Limited range of motion (flexion and extension) of the limbs can make insertion of invasive arterial cannulae and intravenous access difficult. These difficulties may be compounded by anatomical malposition of the vessels. As was noted with our patient, the use of ultrasound guidance may be invaluable to aid in gaining adequate vascular access for major surgical procedures [49, 50]. The muscle wasting and joint contractures also mandate close attention to surgical positioning.
In summary, GA-1 is an autosomal recessive metabolic mitochondrial disorder due to the deficiency of the enzyme, GCDH. Specific perioperative comorbid concerns include the associated underlying seizure disorder and the potential for perioperative respiratory compromise related to respiratory or upper airway involvement. Prolonged fasting may lead to hypoglycemia and increased catabolism resulting in worsening of physiologic homeostasis. Anesthetic concerns include the potential for metabolic derangements from the effects of prolonged propofol infusions on mitochondrial fatty acid metabolism and the potential for prolonged effects of neuromuscular blocking agents. The preoperative assessment of end-organ impairment by the primary disease process and close postoperative monitoring are mandatory for the effective perioperative care of these patients.
| References | ▴Top |
- Goodman SI, Kohlhoff JG. Glutaric aciduria: inherited deficiency of glutaryl-CoA dehydrogenase activity. Biochem Med. 1975;13(2):138-140.
doi - Lindner M, Kolker S, Schulze A, Christensen E, Greenberg CR, Hoffmann GF. Neonatal screening for glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27(6):851-859.
doi pubmed - Heringer J, Boy SP, Ensenauer R, Assmann B, Zschocke J, Harting I, Lucke T, et al. Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol. 2010;68(5):743-752.
doi pubmed - Tang NL, Hui J, Law LK, Lam YY, Chan KY, Yeung WL, Chan AY, et al. Recurrent and novel mutations of GCDH gene in Chinese glutaric acidemia type I families. Hum Mutat. 2000;16(5):446.
doi - Kolker S, Garbade SF, Boy N, Maier EM, Meissner T, Muhlhausen C, Hennermann JB, et al. Decline of acute encephalopathic crises in children with glutaryl-CoA dehydrogenase deficiency identified by newborn screening in Germany. Pediatr Res. 2007;62(3):357-363.
doi pubmed - Kolker S, Koeller DM, Okun JG, Hoffmann GF. Pathomechanisms of neurodegeneration in glutaryl-CoA dehydrogenase deficiency. Ann Neurol. 2004;55(1):7-12.
doi pubmed - Pitt JJ, Eggington M, Kahler SG. Comprehensive screening of urine samples for inborn errors of metabolism by electrospray tandem mass spectrometry. Clin Chem. 2002;48(11):1970-1980.
pubmed - Goodman SI, Markey SP, Moe PG, Miles BS, Teng CC. Glutaric aciduria; a "new" disorder of amino acid metabolism. Biochem Med. 1975;12(1):12-21.
doi - Ozand PT, Gascon GG. Organic acidurias: a review. Part 1. J Child Neurol. 1991;6(3):196-219.
doi pubmed - Mueller P, Schulze A, Schindler I, Ethofer T, Buehrdel P, Ceglarek U. Validation of an ESI-MS/MS screening method for acylcarnitine profiling in urine specimens of neonates, children, adolescents and adults. Clin Chim Acta. 2003;327(1-2):47-57.
doi - Wilcox RL, Nelson CC, Stenzel P, Steiner RD. Postmortem screening for fatty acid oxidation disorders by analysis of Guthrie cards with tandem mass spectrometry in sudden unexpected death in infancy. J Pediatr. 2002;141(6):833-836.
doi pubmed - Martin DP, Bhalla T, Thung A, Rice J, Beebe A, Samora W, Klamar J, et al. A preliminary study of volatile agents or total intravenous anesthesia for neurophysiological monitoring during posterior spinal fusion in adolescents with idiopathic scoliosis. Spine (Phila Pa 1976). 2014;39(22):E1318-1324.
doi pubmed - Zytkovicz TH, Fitzgerald EF, Marsden D, Larson CA, Shih VE, Johnson DM, Strauss AW, et al. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem. 2001;47(11):1945-1955.
pubmed - Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348(23):2304-2312.
doi pubmed - Bjugstad KB, Goodman SI, Freed CR. Age at symptom onset predicts severity of motor impairment and clinical outcome of glutaric acidemia type 1. J Pediatr. 2000;137(5):681-686.
doi pubmed - Poge AP, Autschbach F, Korall H, Trefz FK, Mayatepek E. Early clinical manifestation of glutaric aciduria type I and nephrotic syndrome during the first months of life. Acta Paediatr. 1997;86(10):1144-1147.
doi pubmed - Kolker S, Christensen E, Leonard JV, Greenberg CR, Boneh A, Burlina AB, Burlina AP, et al. Diagnosis and management of glutaric aciduria type I--revised recommendations. J Inherit Metab Dis. 2011;34(3):677-694.
doi pubmed - Kyllerman M, Skjeldal O, Christensen E, Hagberg G, Holme E, Lonnquist T, Skov L, et al. Long-term follow-up, neurological outcome and survival rate in 28 Nordic patients with glutaric aciduria type 1. Eur J Paediatr Neurol. 2004;8(3):121-129.
doi pubmed - Hald JK, Nakstad PH, Skjeldal OH, Stromme P. Bilateral arachnoid cysts of the temporal fossa in four children with glutaric aciduria type I. AJNR Am J Neuroradiol. 1991;12(3):407-409.
pubmed - Martinez-Lage JF, Casas C, Fernandez MA, Puche A, Rodriguez Costa T, Poza M. Macrocephaly, dystonia, and bilateral temporal arachnoid cysts: glutaric aciduria type 1. Childs Nerv Syst. 1994;10(3):198-203.
doi pubmed - Hartley LM, Khwaja OS, Verity CM. Glutaric aciduria type 1 and nonaccidental head injury. Pediatrics. 2001;107(1):174-175.
doi pubmed - Kafil-Hussain NA, Monavari A, Bowell R, Thornton P, Naughten E, O'Keefe M. Ocular findings in glutaric aciduria type 1. J Pediatr Ophthalmol Strabismus. 2000;37(5):289-293.
pubmed - Christensen E. Improved assay of glutaryl-CoA dehydrogenase in cultured cells and liver: application to glutaric aciduria type I. Clin Chim Acta. 1983;129(1):91-97.
doi - Ituk US, Allen TK, Habib AS. The peripartum management of a patient with glutaric aciduria type 1. J Clin Anesth. 2013;25(2):141-145.
doi pubmed - Teng WN, Lin SM, Niu DM, Kuo YM, Chan KH, Sung CS. Anesthetic management of comprehensive dental restoration in a child with glutaric aciduria type 1 using volatile sevoflurane. J Chin Med Assoc. 2014;77(10):548-551.
doi pubmed - Farag E, Argalious M, Narouze S, DeBoer GE, Tome J. The anesthetic management of ventricular septal defect (VSD) repair in a child with mitochondrial cytopathy. Can J Anaesth. 2002;49(9):958-962.
doi pubmed - Hernandez-Palazon J, Sanchez-Rodenas L, Martinez-Lage JF, Collado IC. Anesthetic management in two siblings with glutaric aciduria type 1. Paediatr Anaesth. 2006;16(2):188-191.
doi pubmed - Goktas U, Kati I, Aytekin OC. Management of outpatient anesthesia in an unusually case with glutaric aciduria type-1. Paediatr Anaesth. 2009;19(6):632-633.
doi pubmed - Tsiotou AG, Malisiova A, Bouzelos N, Velegrakis D. The child with glutaric aciduria type I: anesthetic and perioperative management. J Anesth. 2011;25(2):301-304.
doi pubmed - Martyn JA, Richtsfeld M. Succinylcholine-induced hyperkalemia in acquired pathologic states: etiologic factors and molecular mechanisms. Anesthesiology. 2006;104(1):158-169.
doi - Zhang JM, Wang F, Xin Z, Zi TT, Lv H. Treatment of different-aged children under bispectral index monitoring with intravenous anesthesia with propofol and remifentanil. Eur Rev Med Pharmacol Sci. 2015;19(1):64-69.
pubmed - Choudhry DK, Brenn BR. Bispectral index monitoring: a comparison between normal children and children with quadriplegic cerebral palsy. Anesth Analg. 2002;95(6):1582-1585, table of contents.
doi pubmed - Jaber S, Michelet P, Chanques G. Role of non-invasive ventilation (NIV) in the perioperative period. Best Pract Res Clin Anaesthesiol. 2010;24(2):253-265.
doi pubmed - Pelosi P, Jaber S. Non-invasive respiratory support in the perioperative period. Curr Opin Anaesthesiol. 2010;3:2233-2228.
- Fricker RM, Raffelsberger T, Rauch-Shorny S, Finsterer J, Muller-Reible C, Gilly H, Bittner RE. Positive malignant hyperthermia susceptibility in vitro test in a patient with mitochondrial myopathy and myoadenylate deaminase deficiency. Anesthesiology. 2002;97(6):1635-1637.
doi pubmed - Etcharry-Bouyx F, Sangla I, Serratrice G. [Chronic rhabdomyolysis disclosing mitochondriopathy and malignant hyperthermia susceptibility]. Rev Neurol (Paris). 1995;151(10):589-592.
- Footitt EJ, Sinha MD, Raiman JA, Dhawan A, Moganasundram S, Champion MP. Mitochondrial disorders and general anaesthesia: a case series and review. Br J Anaesth. 2008;100(4):436-441.
doi pubmed - Driessen J, Willems S, Dercksen S, Giele J, van der Staak F, Smeitink J. Anesthesia-related morbidity and mortality after surgery for muscle biopsy in children with mitochondrial defects. Paediatr Anaesth. 2007;17(1):16-21.
doi pubmed - Wolf A, Weir P, Segar P, Stone J, Shield J. Impaired fatty acid oxidation in propofol infusion syndrome. Lancet. 2001;357(9256):606-607.
doi - Uezono S, Hotta Y, Takakuwa Y, Ozaki M. Acquired carnitine deficiency: a clinical model for propofol infusion syndrome? Anesthesiology. 2005;103(4):909.
doi pubmed - Fudickar A, Bein B. Propofol infusion syndrome: update of clinical manifestation and pathophysiology. Minerva Anestesiol. 2009;75(5):339-344.
pubmed - Vasile B, Rasulo F, Candiani A, Latronico N. The pathophysiology of propofol infusion syndrome: a simple name for a complex syndrome. Intensive Care Med. 2003;29(9):1417-1425.
doi pubmed - Jones CT, Raman VT, DeVries S, Cole JW, Kelleher KJ, Tobias JD. Optimizing anticonvulsant administration for children before anesthesia: a quality improvement project. Pediatr Neurol. 2014;51(5):632-640.
doi pubmed - Silva MF, Aires CC, Luis PB, Ruiter JP, L IJ, Duran M, Wanders RJ, et al. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: a review. J Inherit Metab Dis. 2008;31(2):205-216.
doi pubmed - Lin CM, Thajeb P. Valproic acid aggravates epilepsy due to MELAS in a patient with an A3243G mutation of mitochondrial DNA. Metab Brain Dis. 2007;22(1):105-109.
doi pubmed - Mastrangelo M, Celato A. Diagnostic work-up and therapeutic options in management of pediatric status epilepticus. World J Pediatr. 2012;8(2):109-115.
doi pubmed - Adachi M, Ikemoto Y, Kubo K, Takuma C. Seizure-like movements during induction of anaesthesia with sevoflurane. Br J Anaesth. 1992;68(2):214-215.
doi pubmed - Terasako K, Ishii S. Postoperative seizure-like activity following sevoflurane anesthesia. Acta Anaesthesiol Scand. 1996;40(8 Pt 1):953-954.
doi pubmed - Doniger SJ, Ishimine P, Fox JC, Kanegaye JT. Randomized controlled trial of ultrasound-guided peripheral intravenous catheter placement versus traditional techniques in difficult-access pediatric patients. Pediatr Emerg Care. 2009;25(3):154-159.
doi pubmed - Costantino TG, Parikh AK, Satz WA, Fojtik JP. Ultrasonography-guided peripheral intravenous access versus traditional approaches in patients with difficult intravenous access. Ann Emerg Med. 2005;46(5):456-461.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.