

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 5, Number 3, March 2014, pages 182-185
Magnetic Resonance Imaging and Clinical Features in Mayer-Rokitansky-Kuster-Hauser Syndrome
Duygu Kara Bozkurta, Murat Bozkurtb, d, Levent Sahinc
aDepartment of Radiology, Kafkas University Medical Faculty, Kars, Turkey
bDepartment of Obstetric and Gynecology, Kafkas University Medical Faculty, Kars, Turkey
cPark Hospital IVF Center, Malatya, Turkey
dCorresponding author: Murat Bozkurt, Department of Obstetric and Gynecology, Kafkas University Medical Faculty, Kars, Turkey
Manuscript accepted for publication January 14, 2014
Short title: Mayer-Rokitansky-Kuster-Hauser Syndrome
doi: https://doi.org/10.14740/jmc1662w
| Abstract | ▴Top |
Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome is a rare syndrome that results from the disruption of the embryonic Mullerian duct development, accompanied with genital and renal malformations. Patients are characterized by partial or complete uterine aplasia with aplasia or hypoplasia of upper 2/3 of the vagina. The aim of this case report is to discuss effectiveness of magnetic resonance imaging and possible treatment options in a 24-year-old patient who was admitted to our clinic complaining primary amenorrhea and diagnosed with MRKH type II syndrome. Accurate diagnosis and evaluation of accompanying other system anomalies are quite important in MRKH syndrome in terms of treatment methods and assisted reproductive techniques.
Keywords: Primary amenorrhea; Mayer-Rokitansky-Kuster-Hauser syndrome; Mullerian defects; Urinary tract anomalies; Magnetic resonance imaging
| Introduction | ▴Top |
Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome is a rare syndrome that results from the disruption of the embryonic Mullerian duct development, accompanied by genital and renal malformations. For the first time, Mayer (1829) and Rokitansky (1838) identified it as a uterine and vaginal agenesis secondary to developmental anomalies of the uterine canal [1]. Kuster defined urological components in MRKH in 1910, and defined differentiation from testicular feminization syndrome in 1961. External genitalia appearance and the development of secondary sex characteristics are normal in adolescence in MRKH syndrome cases. Therefore, the majority of cases are admitted with complaints of primary amenorrhea in adolescent period. Patients who are diagnosed before puberty are generally diagnosed incidentally and these patients are admitted to hospital with other health problems in birth or childhood. Mean age at diagnosis was ranged from 15 to 18 years [2]. The aim of this case report is to present the efficacy of magnetic resonance imaging (MRI), clinical features, treatment methods and assisted reproductive techniques in patients with MRKH syndrome.
| Case Report | ▴Top |
A 24-year-old patient was admitted to our clinic with complaint of primary amenorrhea. According to the patient’s medical history, patient was admitted to the doctor many times and each time oral contraceptive pills have been started or been told that you should wait a little more. It was learned from the patient’s family history that patient’s parents have a consanguineous marriages and there is no other known cases of primary amenorrhea in entire family members. Patient’s mother did not use any medications during pregnancy. Pubertal development had begun at 11 years old and thelarche had begun at 13 years old in patient. The patient did not have any pain. Height was measured 155 cm, and body weight was measured 48 kg in physical examination. Secondary sex characteristics such as breast development and axillary and pubic hair were normal. External genitalia, urethra and vaginal orifice were normal in gynecological examination. Imperforate hymen could not be ruled out because patient did not accept vaginal examination.
Total blood count and biochemistry laboratory values were within normal limits. Estradiol: 82 pg/mL (20-160) pg/L, FSH: 5.48 mIU/mL (2.8-11.3), LH: 4.69 (1.1-11.6) mIU/mL, prolactin: 21.94 ng/mL (1.9-25) and androgen levels (free testosterone: 1.81 pg/mL (0.06-2.57) and 17-hydroxyprogesterone: 0.3 ng/mL (0.10-1.0); androstenedione: 1.8 ng/mL (0 from 0.21 to 3, 08), DHEA-S: 145 g/dL (65-380)) were normal in hormonal evaluation.
Hypoechoic structure which is approximately 18 × 15 × 10 mm in size, behind the bladder and midline in position, and maybe the rudimentary uterus was observed in pelvic ultrasonography. Both ovaries could not be visualized. The left kidney could not be visualized in abdominal ultrasound. There was an ectopic kidney variation on the right side and right kidney was seen in the right half of the pelvis. Pelvic MRI was planned in order to evaluate both ovaries and hypoechoic structure behind the bladder.
Pelvic MRI was performed at 1.5 T (Sonata; Siemens, Erlangen, Germany) MR device by using pelvic phased-array coil. Pelvic MR images were obtained by sagittal T2 turbo spin echo (TSE), axial T2 TSE TRA fat sat, axial pre- and post-contrast T1 TSE TRA fat sat sequence. Approximately 15 × 5 mm in diameter hypointense viewed uterine remnant in retrovesical area was seen in MRI (Fig. 1). Cervix uteri and upper 2/3 of the vagina were not observed. Right ovary could not be visualized. Left ovary was measured 17 × 15 mm in diameter and it was observed above the normal localization secondary to incomplete descent (Fig. 2).
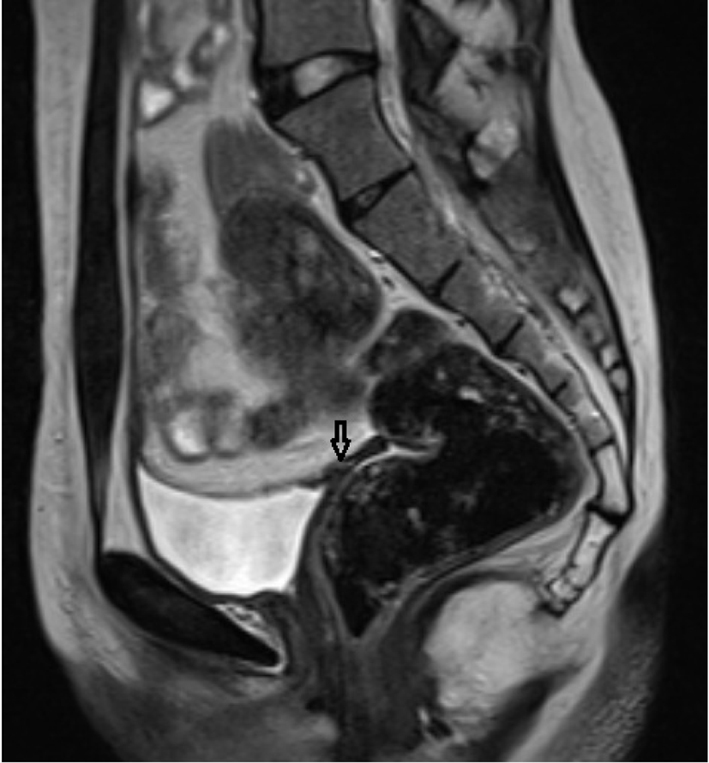 Click for large image | Figure 1. Rudimentary uterus in the retrovesical area in sagittal T2 TSE sequence (arrow). |
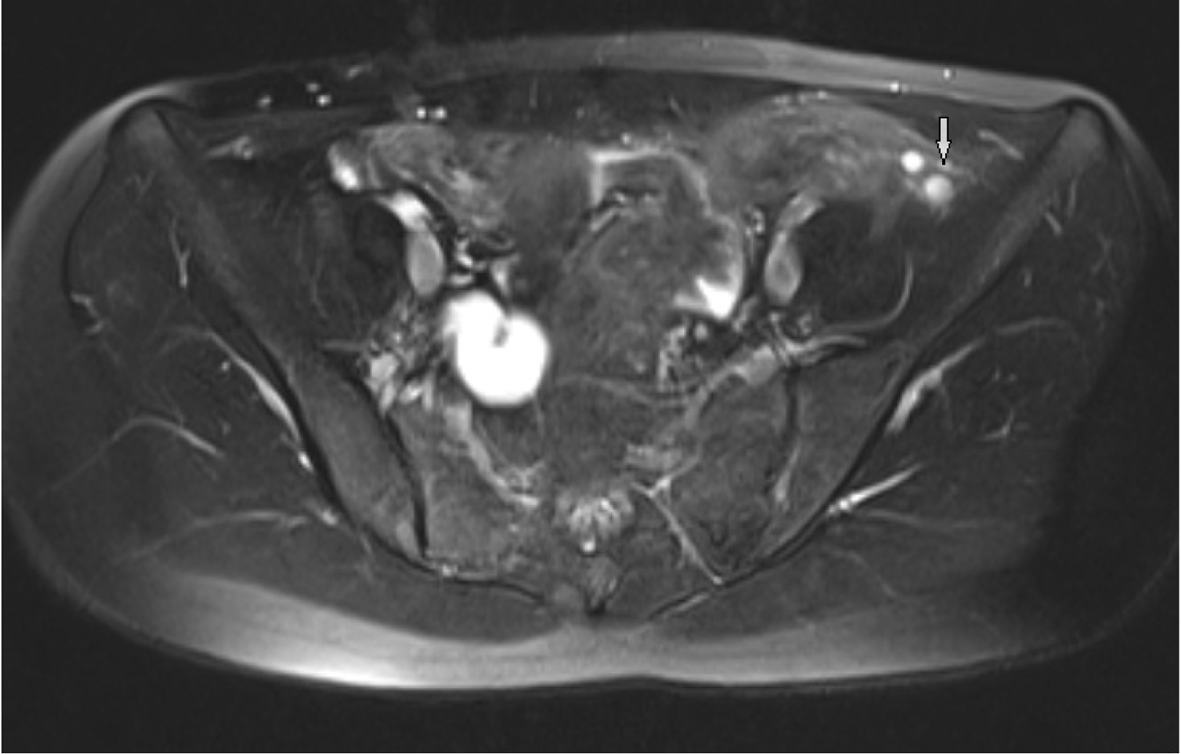 Click for large image | Figure 2. Observing follicles in the ovary secondary to incomplete descent in axial T2 TSE TRA fat sat sequence. |
46, XX chromosome structures have been seen after the karyotype analysis in patient who was suspected to have MRKH syndrome. This result supported the diagnosis of MRKH syndrome. Patient was informed about this situation and psychological support was provided. In addition, patient was informed about the methods of assisted reproduction that can be applied in the future.
| Discussion | ▴Top |
MRKH syndrome is the second most common cause of primary amenorrhea and it is observed in approximately 15% of primary amenorrhea patients [3]. The incidence of the syndrome has been reported as 1:4,000 [4]. Although the reason of MRKH syndrome is not fully understood, it is implicated that polygenic and environmental factors are responsible in etiology. In our case, there were no history about gestational diabetes in mother, and thalidomide or diethylstilbestrol using during pregnancy. There was no other MRKH syndrome in sisters and the other female family members. The first finding that suggests the diagnosis of MRKH syndrome is failure to monitor menarche during puberty in spite of normal development of secondary sex characteristics. Diagnosis would be clear after using radiologic modalities and karyotype analysis.
The female internal genital tract (fallopian tubes, uterus, cervix and 2/3 upper vagina) develops from Mullerian duct during embryogenesis. MRKH syndrome is thought to arise from pausing of the development and differentiation of the Mullerian duct after 7 weeks of embryogenesis.
Urinary tract anomalies can be accompanied with MRKH syndrome because embryologically urinary system and genital system develop together. MRKH syndrome is classified into type I and II according to accompanying urinary and other system abnormalities. Bilateral ovaries, fallopian tubes and renal systems development are normal in type I MRKH syndrome. Complete uterine aplasia or two rudimentary horn associated with peritoneal folds are found. Lower 1/3 portion of vagina can be complete in its development, may be terminated with a blind pouch or may be atresic because it is originated from ectodermal cells. Type II MRKH syndrome may accompany with Mullerian duct aplasia, renal dysplasia and cervical somites anomaly with unilateral renal agenesis, renal ectopia and horseshoe kidney variation, skeletal system anomalies especially vertebral anomalies and scoliosis, hearing problems, heart defects, syndactyly and polydactyly [5]. In our case, there were pelvic kidney variation on the right side and renal agenesis on the left side (Fig. 3). Other systems were normal.
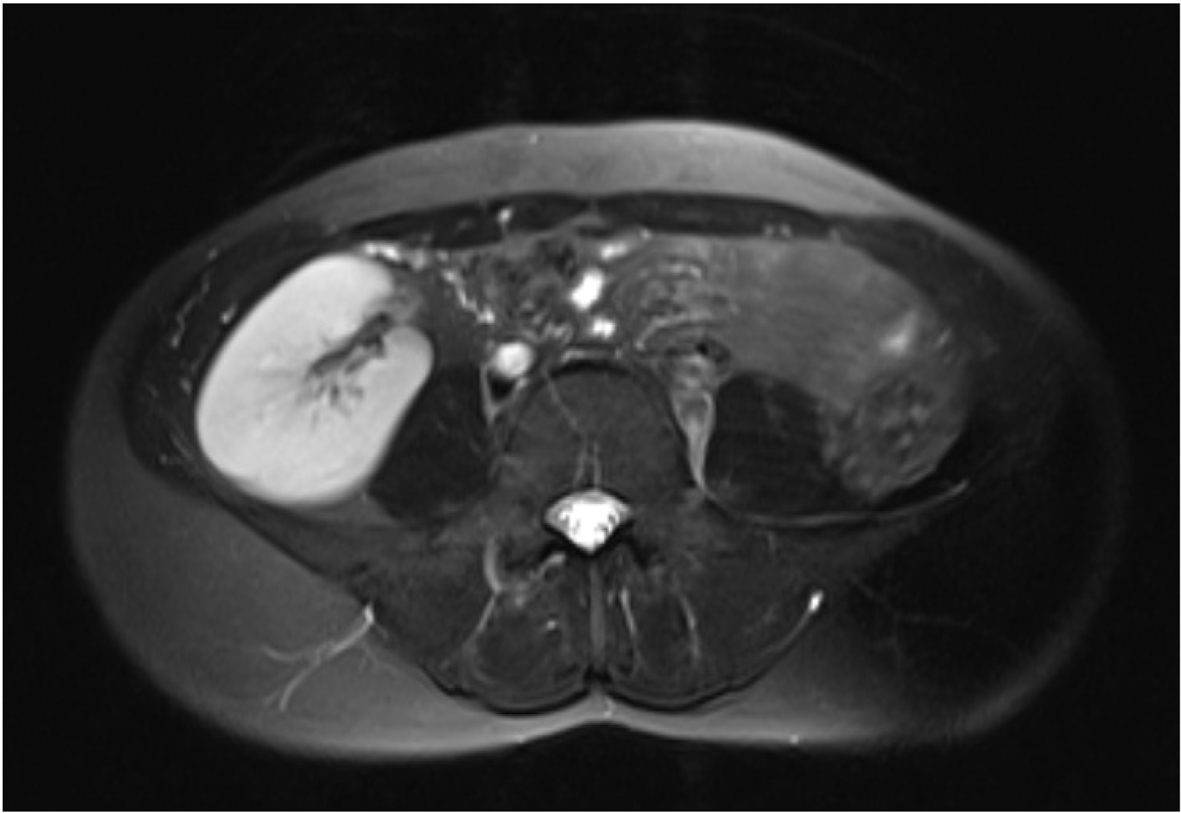 Click for large image | Figure 3. Ectopic kidney localized in the pelvic region. |
Isolated vaginal atresia, WNT4 syndrome, transverse vaginal septum, imperforate hymen and testicular feminization should be considered for the differential diagnosis of MRKH syndrome. In isolated vaginal atresia, the presence of the top 2/3 of vagina is variable, but uterus and ovaries are normally observed. In transverse vaginal septum and imperforate hymen, 2/3 portion of top of the vagina and uterus are normally observed [6]. Ectopic testis tissue is observed instead of ovaries in testicular feminization cases. Therefore, bilateral superficial inguinal USG should be done in order to determine ectopic testis tissue.
Pelvic ultrasonography is the first method of choice in the diagnosis of MRKH because it is simple, inexpensive and non-invasive. It is not always so easy to detect atresic or malformated internal genital organs on ultrasound. Therefore, the experience of the radiologist who performed ultrasonography is very important. In our case, a thin hypoechoic septa was observed in the localization of the uterus. Both ovaries could not be visualized. Because of the inadequacy of ultrasound findings, pelvic MRI was ordered.
MRI which is free of radiation, non-invasive and multiplanar imaging method, has a high soft tissue resolution. Therefore, pelvic anatomy and possible pathological conditions can be shown more clearly. It is quite important to detect rudimentary uterus between bladder and rectum in sagittal T2-weighted sequences, and to detect vaginal atresia with not visualizing normal vagina between rectum and urethra in the axial plane. MRI is also important to detect accompanying other urinary tract and anorectal malformation.
Uterine tissue and the upper 2/3 of the vagina are not clearly indistinguishable while the lower 1/3 of the vagina that developed from different embryonic structure is always observed in MRKH syndrome. Hematometra in various degrees can be observed as a secondary to uterine development and the presence of endometrial glands in approximately 6-10% of patients with MRKH syndrome. This situation can cause cyclic abdominal pain [7].
All information that can be provided from ultrasound, intravenous pyelography and diagnostic laparoscopy, can be obtained with MRI [2]. MRI’s sensitivity and specificity were found 100% compared with laparoscopic findings in terms of diagnosing MRKH syndrome in a study conducted on 56 patients with MRI findings diagnosed with MRKH syndrome [8]. There is no specific MRI protocol for MRKH syndrome. Every center uses different MRI sequences and shot plans in order to evaluate pelvic organs with detail and high resolution.
T1- and T2-weighted sequences must be obtained in the axial, coronal and sagittal plans in pelvic MRI. T2-weighted sequences are very important to determine zonal anatomy of the uterus in female pelvic MRI [9]. Sagittal T2-weighted spin-echo and fast spin-echo sequences are very important in determining uterovaginal anomalies [10]. The diagnosis of uterine hypoplasia and aplasia can be best diagnosed in the sagittal T2-weighted images. The axial images are quite helpful for vaginal hypoplasia and aplasia. The normal vagina has an intermediate signal intensity. The bladder and urethra are located at the anterior of the vagina, anal canal is at the posterior of the vagina [10]. We can not see any structure in this location in vaginal agenesis. In the present study, we used sagittal T2-weighted TSE images for uterovaginal anomalies and rudimantary uterus and also axial fat sat T2 TSE sequences for visualization of the ovarian follicules. The abdominal organs can easily evaluate and the diagnosis of accompanying urinary tract abnormalities can be made by wide field of view sequences in coronal plane. Sagittal T2-weighted sequences are very useful in the diagnosis of hematometrocolpos which develops after transverse vaginal septum and imperforate hymen and are important for the differential diagnosis of MRKH syndrome. It is a very important imaging method to visualize rudimentary ectopic testis in order to distinguish testicular feminization from MRKH syndrome. Failures to observe uterus, upper 2/3 of the vagina and ovarian tissue are highly specific for the diagnosis of MRKH syndrome. Because ovaries are not always in the normal localization, the detection of functional follicles in MRI is an important marker. In our case, right ovary could not be visualized and left ovary was observed above the normal localization secondary to incomplete descent.
There are many surgical methods for treatment [11]. Vaginoplasty surgery can be performed open or closed surgical technique in many ways after recovery of sexual function and emotional development [12]. Today, MRKH syndrome cases cannot have children without surrogate mother. Ovaries respond well to gonadotropin in IVF treatment. Embryo transfer can be made to surrogate mother after combining the father’s sperm and mother’s oocytes. However, this situation can create many legal status and procedure.
As a result, MRI is a useful, easy and non-invasive imaging method and can be used by itself instead of many invasive or non-invasive diagnosing methods in order to diagnose MRKH syndrome and other accompanying system abnormalities.
| References | ▴Top |
- Pittock ST, Babovic-Vuksanovic D, Lteif A. Mayer-Rokitansky-Kuster-Hauser anomaly and its associated malformations. Am J Med Genet A. 2005;135(3):314-316.
doi pubmed - Govindarajan M, Rajan RS, Kalyanpur A, Ravikumar. Magnetic resonance imaging diagnosis of Mayer-Rokitansky-Kuster-Hauser syndrome. J Hum Reprod Sci. 2008;1(2):83-85.
doi pubmed - Strubbe EH, Cremers CW, Willemsen WN, Rolland R, Thijn CJ. The Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome without and with associated features: two separate entities?. Clin Dysmorphol. 1994;3(3):192-199.
pubmed - Oppelt P, Renner SP, Kellermann A, Brucker S, Hauser GA, Ludwig KS, Strissel PL,
et al . Clinical aspects of Mayer-Rokitansky-Kuester-Hauser syndrome: recommendations for clinical diagnosis and staging. Hum Reprod. 2006;21(3):792-797.
doi pubmed - Griffin JE, Edwards C, Madden JD, Harrod MJ, Wilson JD. Congenital absence of the vagina. The Mayer-Rokitansky-Kuster-Hauser syndrome. Ann Intern Med. 1976;85(2):224-236.
doi pubmed - Ustuner I, Keskin L, Ozturk O, Ozyigit E, Avsar AF. Mayer-Rokitansky-Kuster-Hauser (MRKH) sendromu. Yeni Tip Dergisi. 2008;25:241-244.
- Giusti S, Fruzzetti E, Perini D, Fruzzetti F, Giusti P, Bartolozzi C. Diagnosis of a variant of Mayer-Rokitansky-Kuster-Hauser syndrome: useful MRI findings. Abdom Imaging. 2011;36(6):753-755.
doi pubmed - Pompili G, Munari A, Franceschelli G, Flor N, Meroni R, Frontino G, Fedele L,
et al . Magnetic resonance imaging in the preoperative assessment of Mayer-Rokitansky-Kuster-Hauser syndrome. Radiol Med. 2009;114(5):811-826.
doi pubmed - Marcal L, Nothaft MA, Coelho F, Volpato R, Iyer R. Mullerian duct anomalies: MR imaging. Abdom Imaging. 2011;36(6):756-764.
doi pubmed - Saleem SN. MR imaging diagnosis of uterovaginal anomalies: current state of the art. Radiographics. 2003;23(5):e13.
doi pubmed - Pizzo A, Lagana AS, Sturlese E, Retto G, Retto A, De Dominici R, Puzzolo D. Mayer-rokitansky-kuster-hauser syndrome: embryology, genetics and clinical and surgical treatment. ISRN Obstet Gynecol. 2013;2013:628717.
- Jabeen M. Mayer-rokitansky-kuster-hauser syndrome. World Journal of Laparoscopic Surgery. 2011;(4):123-128.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.