

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 13, Number 6, June 2022, pages 302-306

Peutz-Jeghers Syndrome Manifested as Multiple Polyps in Jejunum With Intussusception
Hai Hua Jianga, Feng Lua, Shu Guang Tana, Sai Qi Hea, b
aDepartment of Gastrointestinal Surgery, Hengyang Central Hospital, Hengyang, Hunan 421000, China
bCorresponding Author: Sai Qi He, Department of Gastrointestinal Surgery, Hengyang Central Hospital, Hengyang, Hunan 421000, China
Manuscript submitted April 8, 2022, accepted June 2, 2022, published online June 11, 2022
Short title: Multiple Polyps With Intussusception in PJS
doi: https://doi.org/10.14740/jmc3944
| Abstract | ▴Top |
Peutz-Jeghers syndrome (PJS) is a relatively rare autosomal dominant genetic disease, often manifested as mucous membranes, skin pigmented spots and multiple polyps in the gastrointestinal tract. It can be followed by a variety of serious complications such as bleeding, obstruction, intussusception, and malignant transformation. We introduce the case of a 26-year-old male patient who was diagnosed with multiple polyps in the jejunum with intussusception caused by PJS. He was discharged after emergency surgery reduction and partial resection of the small intestine. Gastrointestinal polyps, hemorrhage, intussusception, intestinal obstruction, and increased risk of cancer occur in patients with PJS. Currently, polypectomy under endoscopic techniques, reexamination and follow-up are the main treatment options; surgical treatment is used for bleeding, intussusception, and cancer. Therefore, it is very necessary for us to have a correct understanding of it, actively prevent it, treat it and follow these patients closely.
Keywords: Peutz-Jeghers syndrome; Mucosal pigmentation; Intestinal polyps; Intussusception; Treatment
| Introduction | ▴Top |
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant genetic disease caused by LKB1/STK11 germline mutation [1, 2]. The syndrome was first described by Peutz in 1921. In 1949, Jeghers et al introduced the relevant clinical features of the disease in detail and systematically [3]. In 1954, Dr. Bruwer of the Mayo Clinic named the syndrome as “Peutz-Jeghers syndrome”, with an incidence of 1/200,000 to 1/50,000 [1, 2, 4, 5]. PJS mainly has the following three clinical features: 1) Melanoma that can be seen on the lips, nose, cheeks and the palms of the extremities, and in a few patients it can also be found on the palm and sole surface, eyelids, tongue, perineal skin and gastrointestinal mucosa; 2) Gastrointestinal polyps that can be found in any part of the gastrointestinal tract, mainly in the small intestine; 3) Genetic features: it is an autosomal dominant genetic disorder. Studies have shown that mutations in the 3p14 region and STK11 gene are closely related to the pathogenesis of PJS [6]. Since it can be complicated by a variety of serious complications such as gastrointestinal bleeding, intestinal obstruction, intussusception, and malignant transformation, PJS seriously affects the quality of life of patients. Therefore, it is very necessary for us to have a correct understanding of it, actively prevent and treat it, and follow these patients closely. Some studies have reported [7] that active endoscopic polypectomy has positive significance for preventing polyp-induced obstruction, bleeding, perforation, and malignant transformation, and reduces related complications and patient suffering. At present, the clinical treatment of this disease is mainly to solve the acute abdomen caused by PJS, such as intestinal obstruction, intussusception, hemorrhage, and complications such as malignant transformation.
| Case Report | ▴Top |
Investigations
A 26-year-old male presented to the Emergency Department of our hospital complaining of “abdominal pain for 7 h”. The patient had intermittent abdominal pain for more than half a year, and the abdominal pain worsened 7 h ago, accompanied by vomiting, with no blood in the stool, and no history of weight loss. The patient had been diagnosed with PJS in childhood (diagnosed in other hospitals, specific treatment unknown). In the past, open bowel polypectomy was performed for intestinal polyps in another hospital. Later, he complained that he underwent surgery for intestinal prolapse (with the specific name of the disease and the specific surgical method unknown). His mother and sister suffered from PJS and mother died of intestinal malignancy. His vital signs were normal. Mucocutaneous pigmentation was noted on his lips, palms, and soles of the feet (Fig. 1). Tension of abdominal muscles, tenderness of the whole abdomen, the tenderness around the umbilicus and the left lower quadrant are more obvious, and bowel sounds are significantly reduced.
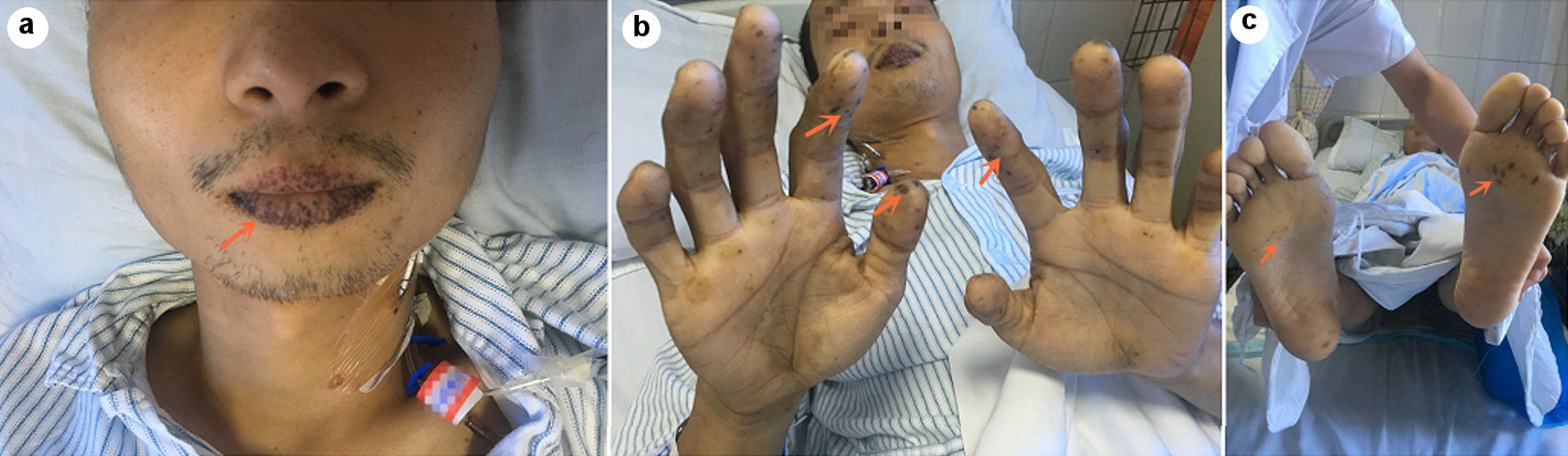 Click for large image | Figure 1. Melanin spots (arrows) were seen on the patient’s lips (a), palms of fingers (b) and soles of feet (c). |
Diagnosis
Mucocutaneous pigmentation was noted on his lips, palms, and soles of the feet. Computed tomography (CT) of the abdomen showed that the small intestine of about groups 4 and 5 in the left middle and lower abdominal region having concentric circles changes; and the intestinal wall was obviously edematous and widened (Fig. 2a); and slightly high-density shadow changes can be seen in the bowel (Fig. 2b). Based on the past history, clinical manifestations, and CT imaging examination, a diagnosis of PJS with multiple polyps in jejunum with intussusception was made.
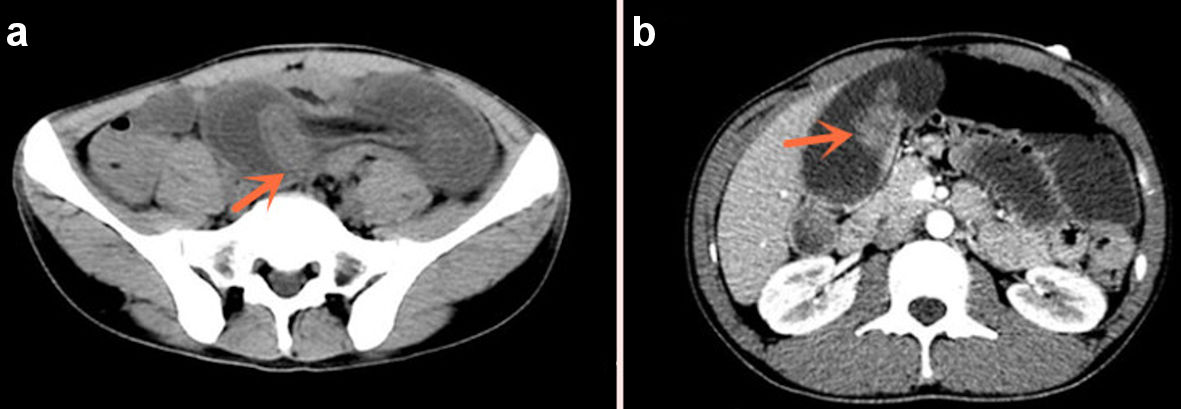 Click for large image | Figure 2. Concentric changes and obstruction of the bowel (a), high-density mass in the bowel (b) (arrows). |
Treatment
The patient underwent emergency exploratory laparotomy. The exploration showed: about 300 mL of pale red exudate in the abdominal cavity, about 150 cm away from the ligament of Treitz, and the distal jejunum is inserted into the proximal jejunum (Fig. 3a), forming a length of about 40-cm jejunum-jejunum intussusception, part of the bowel showed congestion and swelling changes, and the proximal jejunum was obviously dilated. We performed the intussusception reposition, and found multiple lumps in the dilated bowel, the size of the lump is about 1.5 - 2.5 cm (Fig. 3b). Considering the jejunal mass and intussusception, it was decided to perform partial jejunectomy and side-to-side anastomosis of the small intestine. Several polypoid masses were found in the excised bowel (Fig. 3c), with a maximum size of about 1.5 × 1.5 cm and soft texture. Postoperative pathological immunohistochemistry showed: jejunum Peutz-Jeghers polyps, which were multiple (Fig. 4).
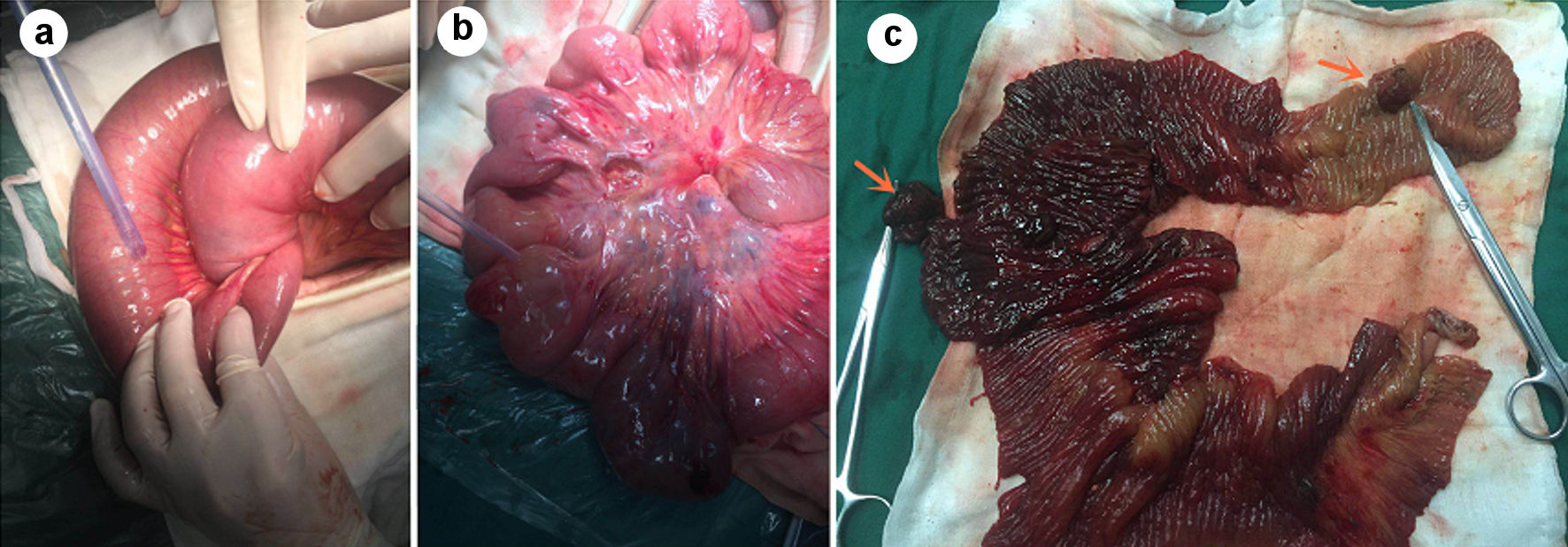 Click for large image | Figure 3. Jejunum-jejunum intussusception and obstruction of the intestinal segment (a), multiple mass-like changes in the intestinal tract (b), several polypoid tumors, the largest size is about 1.5 × 1.5 cm, the texture is soft and pedunculated (c) (shown by arrow). |
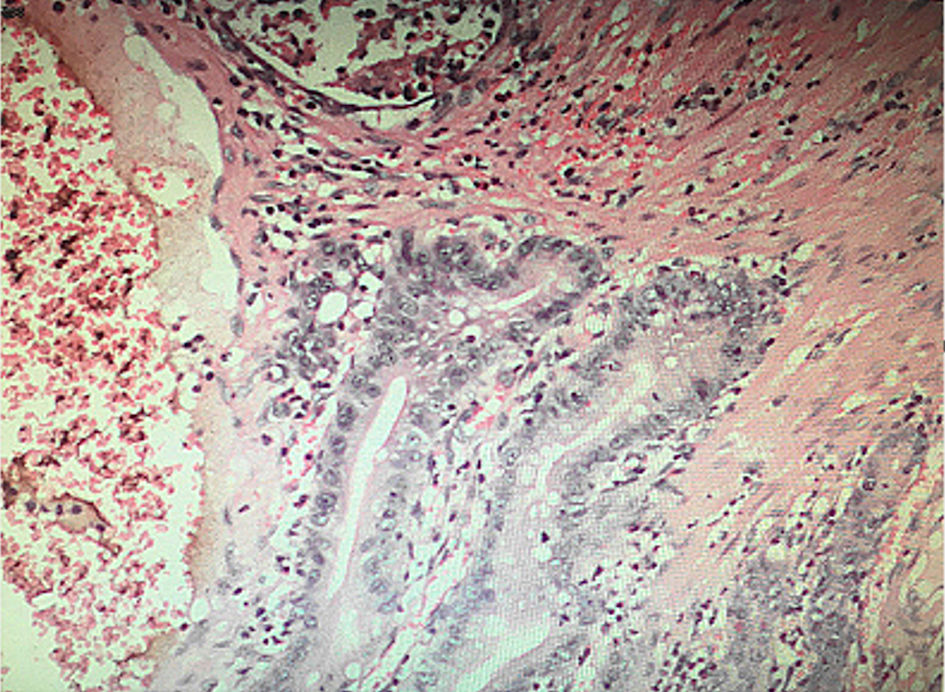 Click for large image | Figure 4. Pathological immunohistochemical indication. The central smooth muscle showed tree branching pattern changes, and glands can be seen in the surface mucosa. |
Follow-up and outcomes
The postoperative recovery was well, and the follow-up was good 1 month after discharge.
| Discussion | ▴Top |
PJS is also called “dark spot-polyposis syndrome” because of its clinical manifestations of the lips and finger end skin and mucous membranes accompanied by melanotic plaque changes and accompanied by multiple polyposis changes in the digestive tract. The disease was first reported by Peutz in 1921 and was systematically described by Jeghers et al in 1949, so it was named “Peutz-Jeghers syndrome”. It is more common in children and adolescents and is autosomal dominant [8]. Some studies have reported that about 30% are inherited in families, and 70% may be caused by gene mutation [9]. Studies have shown that among the many causes of PJS, its genes are mainly related to the mutation of a susceptibility gene called STK11/LKB1 on chromosome 19p13.3 [6]. About 66-94% of genetic deletions occur in chromosome 19 (19p13.3) on the broken arm of LKB1/STK11 gene [1, 2]. Among them, Chen et al [10] found in the comparison of familial and sporadic cases of PJS, the LKB1 gene mutation rate was as high as 85.7% in familial PJS, and it was 63.2% in all PJS patients. Some studies have confirmed the LKB1/STK11 mutation in hereditary colorectal tumor-related genes in polyp tissue of PJS patients [11]. Therefore, looking at the current research reports on PJS, it is indicated that LKB1 gene mutation is the main factor leading to the occurrence of familial PJS. In terms of genetic disease, whether PJS is caused by a single gene mutation, or a combination of multiple gene mutations remains to be further studied.
Among PJS patients, about 95% of them have skin and mucous membrane melanoma changes, most of whom are young and adolescents. Mostly, it is scattered in the skin, mucous membranes, fingers, toes, and other parts. Melanin plaques often appear on the face, nose, lips, and oral mucosa, and can also occur on the eyelids and anus. After some patients grow to puberty, a small number of melanin spots gradually fade away. At present, there is no evidence to suggest that this may have a tendency to become cancerous [12].
Another important feature of PJS is polyps, which can be found anywhere in the gastrointestinal tract. The jejunum and ileum polyps are the most common, accounting for 60% to 90% of PJS polyps, followed by colorectal polyps, about 50% to 64%. In addition, there are a small number of clinical cases and literatures showing that PJS patients also show polyp characteristics in some organs other than the gastrointestinal tract, such as the gallbladder, nasal cavity, bronchus, bladder mucosa and ureter mucosa, etc. [13-16]. In terms of histopathological characteristics, polyps are mainly divided into lipomas, hamartomas, inflammatory polyps, hyperplastic polyps, adenoma polyps, etc. [8]. Among them, most of the polyps in PJS patients are hamartoma polyps, and they may coexist with various polyps. A few studies have reported that a small number of PJS polyps manifested as adenomatous polyps alone in pathological features. From this we can see that PJS polyps are mainly manifested as hamartoma and adenomatous polyps, and other types of polyps are relatively rare. Because the PJS polyps are mainly hamartoma polyps, their malignant transformation rate is relatively low, but there are also adenomatous polyps, which lead to a relatively increased risk of cancerous transformation. In addition, some studies have found [17] that hamartoma polyps themselves may gradually transform into cancer under a certain mechanism. In the process of disease evolution, hamartoma gradually develops into adenoma and finally into cancer. Other studies have shown that [18], PJS not only has the above-mentioned carcinogenesis mechanism, but also has a carcinogenesis pathway called de novo mechanism, which is mainly used to explain the carcinogenesis and development of other organs other than gastrointestinal-related malignant tumors. Relevant studies have reported [13, 19] that the risk of cancer in patients with PJS is 15 times that of normal people, and the risks of gastrointestinal tumors are 39% for colon cancer, 29% for gastric cancer, 36% for pancreatic cancer, and 0.5% for esophageal cancer; and there is evidence showing that the average age of cancer in most patients is about 40 years old.
Most PJS patients were admitted to hospital because of abdominal pain, intussusception, and obstruction as the first symptoms; and most of them were treated by emergency surgery. For some asymptomatic gastrointestinal polyps, the diagnosis is mainly based on medical history, family history, physical examination (characteristics of melanin spots), combined with gastrointestinal endoscopy, gastrointestinal imaging, and histopathology. In this case report, the patient underwent emergency surgery for abdominal pain and acute abdomen due to intussusception obstruction. Multiple scattered polyp lesions of the small intestine were found during the operation, and finally PJS was confirmed according to the histopathological results. Reviewing the literature on the treatment of PJS, the focus of clinical treatment of PJS is mainly on the early treatment of gastrointestinal polyposis. Some studies [7, 20] have found that, for the treatment of PJS gastrointestinal polyps, a comprehensive system of endoscopic techniques, close follow-up, combining surgery and preventive drugs is an effective diagnosis and treatment plan. Among them, endoscopic polyp resection is the main method, and for those with scattered polyps, endoscopic resection can be performed in different times. Some studies have reported [21] that double-balloon enteroscopy is of great value and significance in the diagnosis and treatment of PJS. It not only allows the inspection of the entire small bowel, but also facilitates treatment such as microscopic resection, thereby avoiding some surgical resection of the bowel. In the face of difficult endoscopic resection of polyps, malignant transformation, and acute abdomen in patients with PJS, surgical treatment still has an irreplaceable role. On the other hand, from the perspective of genetics, as PJS is a dominant hereditary disease, due to its own particularity, both endoscopic and surgical treatments are still limited treatments in the treatment of PJS. In recent years, with the deepening of domestic and foreign research on PJS, Rossi et al [22] found that they established a PJS nude mouse model in animal experiments and found that the expression of cyclooxygenase-2 (COX-2) in the nude mouse model was increased. At the same time, COX-2 is also highly expressed in hamartoma polyps in PJS patients. Another study found [23, 24] that the application of COX-2 inhibitor drugs (celecoxib) can significantly reduce the incidence of tumor in animals, and to a certain extent reduce the incidence of polyps in patients with PJS. Therefore, COX-2 can be used as a potential new target for the treatment of PJS polyps and PJS syndrome, but further research is needed.
Through the case report, we have a certain understanding of the clinical manifestations, pathogenic genes, pathogenesis, and treatment of PJS. However, its specific pathogenesis has not been fully elucidated, and further research on its mechanism and genetics is required, in order to provide better solutions for clinical diagnosis and treatment of PJS. At present, the treatment of PJS is still mainly endoscopic treatment, with surgical intervention if necessary. We believe that in the clinical diagnosis and treatment of PJS patients in the future, gene therapy may become the dominant in order to achieve the purpose of cure.
Learning points
PJS is a rare autosomal dominant genetic disease. Through this case, we have a certain understanding of its clinical manifestations, pathogenic genes and treatments. Considering the subsequent catastrophic complications, endoscopic examination and treatment should be actively recommended for such patients in clinical practice. Active prevention and close follow-up can improve the quality of life of PJS-patients.
Acknowledgments
We would like to thank Dr. Gao Zhou and Dr. Shui Ping Tang for their supports and all staff of the Department of Gastrointestinal Surgery of Hengyang Central Hospital involved in the care of the patient.
Financial Disclosure
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The authors declare that there are no competing interests.
Informed Consent
The patient authorized the main author, in written form, to publish this case report and all case data.
Author Contributions
Hai Hua Jiang wrote the paper. Sai Qi He managed the case and Feng Lu and Shu Guang Tan analyzed the CT and participated in surgery. All authors read and approved the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Shorning BY, Clarke AR. Energy sensing and cancer: LKB1 function and lessons learnt from Peutz-Jeghers syndrome. Semin Cell Dev Biol. 2016;52:21-29.
doi pubmed - Borun P, De Rosa M, Nedoszytko B, Walkowiak J, Plawski A. Specific Alu elements involved in a significant percentage of copy number variations of the STK11 gene in patients with Peutz-Jeghers syndrome. Fam Cancer. 2015;14(3):455-461.
doi pubmed - Jeghers H, Mc KV, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241(25):993, illust; passim.
doi pubmed - Klimkowski S, Ibrahim M, Ibarra Rovira JJ, Elshikh M, Javadi S, Klekers AR, Abusaif AA, et al. Peutz-Jeghers syndrome and the role of imaging: pathophysiology, diagnosis, and associated cancers. Cancers (Basel). 2021;13(20):5121.
doi pubmed - Stojcev Z, Borun P, Hermann J, et al. Hamartomatous polyposis syndromes. Hereditary Cancer in Clinical Practice. 2013;11(1):779-817.
doi pubmed - Aretz S, Stienen D, Uhlhaas S, Loff S, Back W, Pagenstecher C, McLeod DR, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005;26(6):513-519.
doi pubmed - Mueller CL, Soulellis CA, Kwan J, et al. Sp702 combined surgical and endoscopic clearance of small bowel polyps for Peutz-Jegher's syndrome. Gastrointestinal Endoscopy. 2013;77(5):AB117-AB117.
doi - Shaco-Levy R, Jasperson KW, Martin K, Samadder NJ, Burt RW, Ying J, Bronner MP. Morphologic characterization of hamartomatous gastrointestinal polyps in Cowden syndrome, Peutz-Jeghers syndrome, and juvenile polyposis syndrome. Hum Pathol. 2016;49:39-48.
doi pubmed - Wang HH, Xie NN, Li QY, Hu YQ, Ren JL, Guleng B. Exome sequencing revealed novel germline mutations in Chinese Peutz-Jeghers syndrome patients. Dig Dis Sci. 2014;59(1):64-71.
doi pubmed - Chen C, Zhang X, Wang D, Wang F, Pan J, Wang Z, Liu C, et al. Genetic screening and analysis of LKB1 gene in chinese patients with Peutz-Jeghers syndrome. Med Sci Monit. 2016;22:3628-3640.
doi pubmed - Zhang Z, Duan FX, Gu GL, Yu PF. Mutation analysis of related genes in hamartoma polyp tissue of Peutz-Jeghers syndrome. World J Gastroenterol. 2020;26(16):1926-1937.
doi pubmed - Amos CI, Keitheri-Cheteri MB, Sabripour M, Wei C, McGarrity TJ, Seldin MF, Nations L, et al. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet. 2004;41(5):327-333.
doi pubmed - Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, Cruz-Correa M, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447-1453.
doi pubmed - Ozer A, Sarkut P, Ozturk E, Yilmazlar T. Jejunoduodenal intussusception caused by a solitary polyp in a woman with Peutz-Jeghers syndrome: a case report. J Med Case Rep. 2014;8:13.
doi pubmed - Karaosmanoglu AD, Blake M. Hamartomatous polyp of the gallbladder with an associated choledochal cyst. J Ultrasound Med. 2010;29(11):1663-1666.
doi pubmed - Lim SR, Lee KR, Jeong HK, Kim HI, Cho S, Lee W, Joo Y. A case of Peutz-Jeghers syndrome with nasal polyposis. Korean J Intern Med. 2011;81:630-635.
- Li T, Lin W, Zhao Y, Zhu J, Sun T, Ren L. Distinct promoter methylation patterns of LKB1 in the hamartomatous polyps of Peutz-Jeghers syndrome and its potential in gastrointestinal malignancy prediction. Orphanet J Rare Dis. 2020;15(1):208.
doi pubmed - Zhao ZY, Jiang YL, Li BR, Yang F, Li J, Jin XW, Ning SB, et al. Sanger sequencing in exonic regions of STK11 gene uncovers a novel de-novo germline mutation (c.962_963delCC) associated with Peutz-Jeghers syndrome and elevated cancer risk: case report of a Chinese patient. BMC Med Genet. 2017;18(1):130.
doi pubmed - van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105(6):1258-1264; author reply 1265.
doi pubmed - Wang SL, Gu GL, Wei XM, et al. Clinical comprehensive therapy of Peutz-Jeghers syndrome (report of 71 cases). Chin J Bases Clin General Surg. 2012;19(5):502-506.
- Nakayama Y, Kato S, Kurasawa S, et al. Urgent double-balloon enteroscopy for reduction of jejuno-jejunal intussusception and polypectomy in Peutz-Jeghers syndrome. Journal of Pediatric Surgery Case Reports. 2020;59:101519.
doi - Rossi DJ, Ylikorkala A, Korsisaari N, Salovaara R, Luukko K, Launonen V, Henkemeyer M, et al. Induction of cyclooxygenase-2 in a mouse model of Peutz-Jeghers polyposis. Proc Natl Acad Sci U S A. 2002;99(19):12327-12332.
doi pubmed - McGarrity TJ, Peiffer LP, Amos CI, Frazier ML, Ward MG, Howett MK. Overexpression of cyclooxygenase 2 in hamartomatous polyps of Peutz-Jeghers syndrome. Am J Gastroenterol. 2003;98(3):671-678.
doi pubmed - Udd L, Katajisto P, Rossi DJ, Lepisto A, Lahesmaa AM, Ylikorkala A, Jarvinen HJ, et al. Suppression of Peutz-Jeghers polyposis by inhibition of cyclooxygenase-2. Gastroenterology. 2004;127(4):1030-1037.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.