

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 12, Number 7, July 2021, pages 284-287
Hyperdynamic Valve: A Rare Case of Ehlers-Danlos Syndrome
Sofia Xavier Piresa, c ![]() , Gabriela Pereiraa, David Paivaa, Filipa Cordeirob, Magda Fernandesa, Jorge Cottera
, Gabriela Pereiraa, David Paivaa, Filipa Cordeirob, Magda Fernandesa, Jorge Cottera
aDepartment of Internal Medicine, Hospital Senhora da Oliveira, Guimaraes, Portugal
bDepartment of Cardiology, Hospital Senhora da Oliveira, Guimaraes, Portugal
cCorresponding Author: Sofia Xavier Pires, Hospital Senhora da Oliveira, Rua dos Cutileiros 114, Creixomil, 4835-044 Guimaraes, Portugal
Manuscript submitted March 9, 2021, accepted March 23, 2021, published online May 13, 2021
Short title: Hyperdynamic Valve
doi: https://doi.org/10.14740/jmc3692
| Abstract | ▴Top |
Ehlers-Danlos syndrome is a group of rare genetic disorders of collagen characterized by skin hyperextensibility, joint hypermobility and tissue fragility. The authors describe a rare case of a 52-year-old woman that presented to the clinic with chronic joint pain and talipes equinovarum since childhood. Large eyes, sunken cheeks, thin nose and lobeless ears were noticed on clinical examination. Beighton joint hypermotility criteria were met with a positive Walker and Steinberg sign, elbow extension superior to 10° and knee extension in genu recurvatum more than 10°. An aortic diastolic grade III/VI heart murmur was heard. The complementary study was unremarkable. Moderate aortic insufficiency was found on transthoracic echocardiogram. Genetic testing confirmed positivity for COL1A2, a gene that encodes pro-alpha2 chain type of collagen, which causes cardiac-valvular Ehlers-Danlos syndrome. Authors intend to warn to collagen-related syndromes, since severe complications are associated with a reduced life expectancy for individuals with this condition.
Keywords: Hypermotility; Ehlers-Danlos syndrome; Cardiac-valvular subtype; COL1A2
| Introduction | ▴Top |
Ehlers-Danlos syndrome (EDS) is a heterogeneous group of heritable connective tissue disorders characterized by skin hyperextensibility, joint hypermobility, easy bruising and tissue fragility [1]. Six major types of this disease are characterized: the classical, the hypermobile, vascular, kyphoscoliotic, arthrochalasis and dermatosparaxis. Several mutations in genes encoding fibrillar collagen proteins have been described. These proteins provide structure to connective tissue, with utter importance on skin, parenchymatous organs and blood vessels [2]. Common EDS features such as easy bruising, thin translucent skin, facial features and scarring are usually present in patients with classical forms of EDS but may be absent in some subgroups such as the vascular type. One of the rarest variants, the cardiac-valvular type, is a subgroup with reminiscent of the vascular type, caused by specific recessive variants in the gene encoding pro-alpha2 chain type of collagen (COL1A2) [3]. This syndrome is mainly characterized by generalized joint hypermobility, moderate to severe cardiac-valvular disease and bilateral flatfeet with hindfoot pronation.
Commonly the presentation of the disease is associated with progressive life-threatening complications such as valvular rupture with heart failure, as well as aneurismatic dilation of aortic root. In EDS, the average age for the first major complication is 20 years and more than 80% had had at least one complication by the age of 40. Because of these severe complications, mean life expectancy is shortened to 48 years [2].
| Case Report | ▴Top |
The authors describe a rare case of a 52-year-old woman. She presented to the internal medicine clinic for management of chronic mechanic joint pain associated of small and medium severe joint deformity as well as talipes equinovarum known since childhood. The patient did not report joint redness or swelling, photosensitivity, oral ulcers, red eye, nor she reported precordial pain, irregular beats, or dyspnea. She denied limb paraesthesia, myalgia, or loss of muscle strength.
She had previously consulted with several orthopaedic surgery experts and supportive care with pain management was proposed. The patient was nullipara and past medical history was unremarkable. After eliciting a detailed medical profile, she had family history of a brother with same facial and joint features. No consanguinity was known.
On clinical examination thin velvety translucent skin, large eyes, sunken cheeks, thin nose and lobeless ears, atrophic scars nonrelated to trauma and easy bruising were noticed (Fig. 1). Beighton joint hypermotility criteria were met with a positive Walker and Steinberg sign, elbow extension superior to 10°, and knee extension in genu recurvatum more than 10°. Neurologic examination was unremarkable (Fig. 2). An aortic diastolic grade III/VI heart murmur was heard. An ophthalmology visit did not reveal any signs of crystallin subluxation.
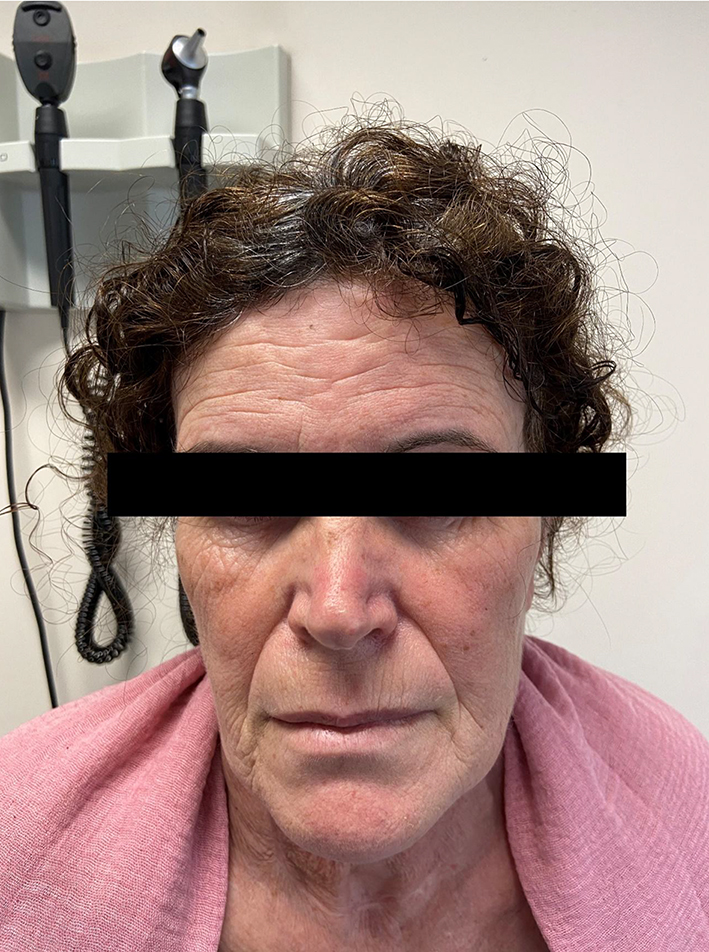 Click for large image | Figure 1. Characteristic features of EDS showing velvety translucent skin, sunken cheeks, thin nose and atrophic scars. EDS: Ehlers-Danlos syndrome. |
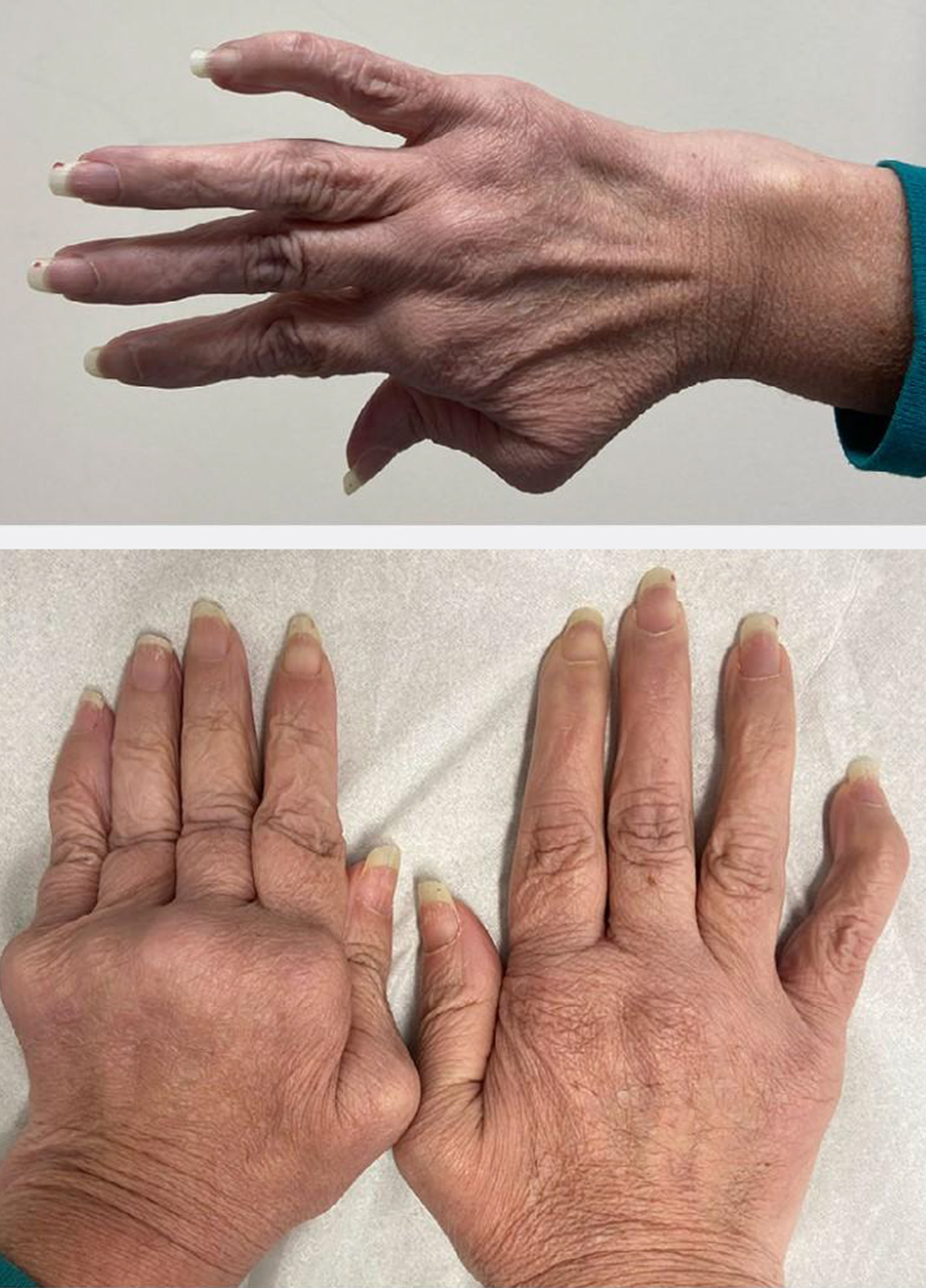 Click for large image | Figure 2. Hypermotility of joint meeting Beighton criteria. |
The complementary study, which included a broad autoimmunity panel, was unremarkable. Moderate aortic regurgitation and mitral valve posterior leaflet billowing was found on transthoracic echocardiogram (Fig. 3). Joint echography revealed subfalangic subluxation.
At this point, a presumption diagnosis of collagen-related disease was proposed. She was asked to avoid hard labor and weight lift since cardiac-valvular Ehlers-Danlos syndrome (cvEDS) was suspected.
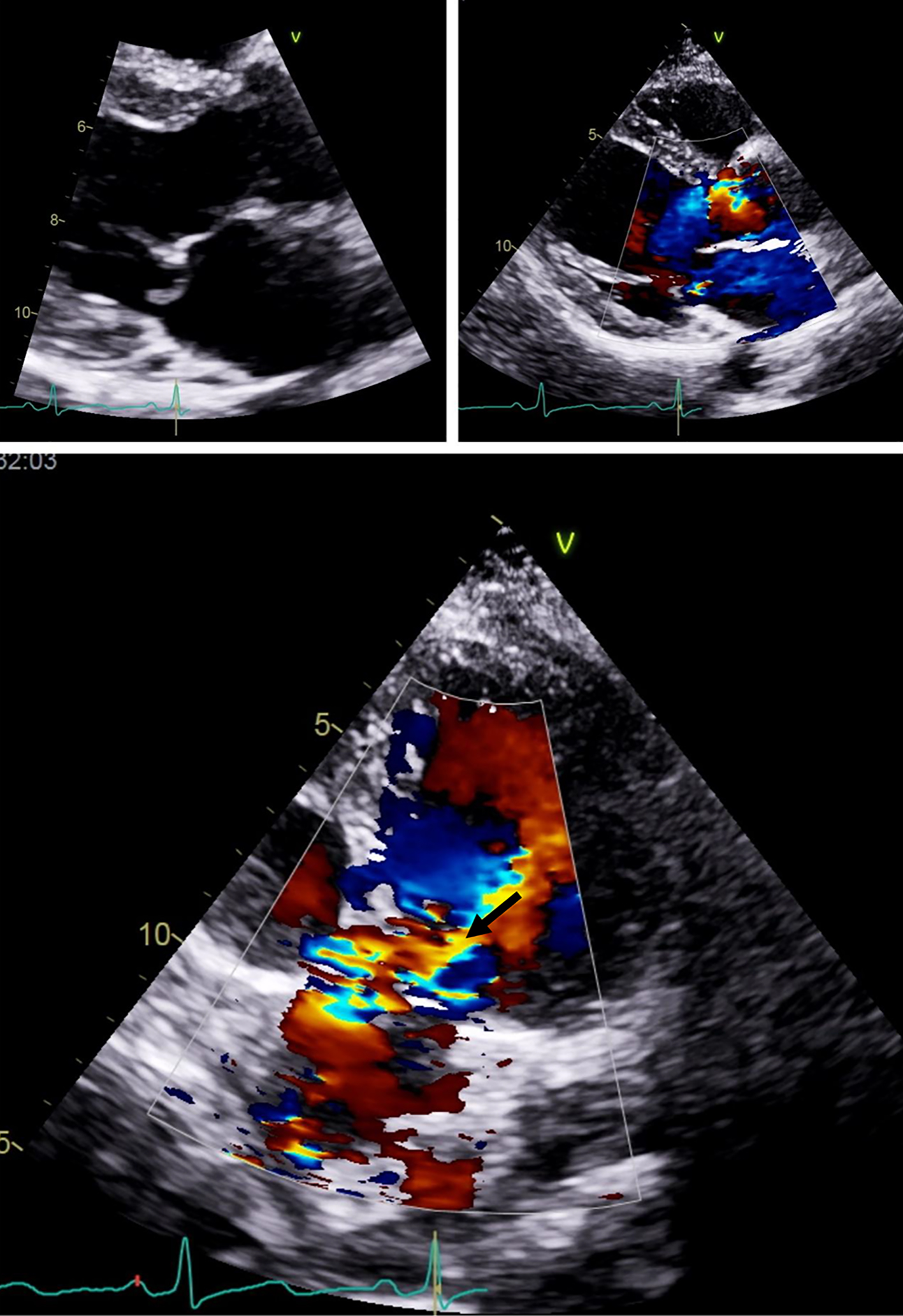 Click for large image | Figure 3. Billowing of posterior leaflet of aortic valve seen on transthoracic echocardiogram (arrow). |
Genetic counselling and testing confirmed positivity for COL1A2, a rare autosomal recessive gene that encodes a cardiac-valvular variant of EDS. Since this variant is highly related to valvular disease, but may also present with aneurisms, artery aneurismatic dilation screening was promptly made with computed tomography anigography of supra-aortic branches, thoracic and abdominal arteries, with no specific findings.
As no treatment is known to date as being effective, pain management was optimized, rehabilitation treatment was prescribed and screening for valvular worsening meeting surgical criteria with close outpatient follow-up was proposed. Appropriate guidance to the patient and family was provided and psychological support was initiated.
| Discussion | ▴Top |
Cardiac-valvular subtype is one of the rarest forms of EDS. We describe here the clinical case of a patient with cvEDS with typical phenotypic features, including hindfoot and talipes equinovarum. Family criteria were met since her brother presented with the same features. Joint hypermotility criteria with more than 12 joints affected were also met. Genetic testing showed a very infrequent recessive mutation on COL1A2 gene, encoding collagen type 1 subunit frequently associated with valvular variant of EDS. No consanguinity was known, but the patient was born in a small, isolated village.
Our case illustrates an awareness of the clinical features of cardiac-valvular type of EDS, since the typical appearance features might not be present. In the presence of articular hypermotility, valvular alterations should be sought, and if present collagen-related disease might be suspected since patients with EDS have a high rate of complications [4] and surgical interventions should be avoided [5] as much as possible.
Learning points
EDS is a heterogeneous group of rare genetic disorders of collagen characterized by skin hyperextensibility, joint hypermobility and tissue fragility.
Most forms of EDS are inherited in an autosomal dominant manner, and a vast array of mutations are known to date.
Cardiac-valvular subtype of EDS is one of the rarest forms of the disease, being inherited in an autosomal recessive manner, and frequently associated with COL1A2 mutation.
Cardiac-valvular subtype of EDS is associated with valvular hypermotility, severe valvular damage and aortic root aneurism.
Screening of valvular alterations is mandatory in patients with articular hypermobility syndromes.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained.
Author Contributions
Sofia Xavier Pires as first author was leader of the medical team and responsible for the diagnosis, data analysis and medical writing. Gabriela Pereira and David Paiva were responsible for data collection and interpretation. Filipa Cordeiro was responsible for cardiologic evaluation of the patient and provided interpretation of transthoracic echocardiogram studies. Magda Fernandes and Jorge Cotter provided senior authorship and critical revision of this article.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Bergqvist D, Bjorck M, Wanhainen A. Treatment of vascular Ehlers-Danlos syndrome: a systematic review. Ann Surg. 2013;258(2):257-261.
doi pubmed - Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342(10):673-680.
doi pubmed - Brooke BS, Arnaoutakis G, McDonnell NB, Black JH, 3rd. Contemporary management of vascular complications associated with Ehlers-Danlos syndrome. J Vasc Surg. 2010;51(1):131-138; discussion 138-139.
doi pubmed - Eagleton MJ. Arterial complications of vascular Ehlers-Danlos syndrome. J Vasc Surg. 2016;64(6):1869-1880.
doi pubmed - Malfait F. Vascular aspects of the Ehlers-Danlos Syndromes. Matrix Biol. 2018;71-72:380-395.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.