

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 2, Number 4, August 2011, pages 139-142
Rapidly Progressive Creutzfeldt-Jakob Disease VV1 Subtype
Anuradha Kollurua, b, Alberto M. Jacira, Mamta Sharmaa
aSt. John Hospital and Medical Center, Detroit, MI, USA
bCorresponding author: 22101 Moross Road, 2nd Floor VEP, Cardiac Cath Lab, St. John Hospital and Medical Center, Detroit, MI, USA
Manuscript accepted for publication May 25, 2011
Short title: Creutzfeldt-Jakob disease
doi: https://doi.org/10.4021/jmc221w
| Abstract | ▴Top |
Creutzfeldt-Jakob disease (CJD) is rare transmissible spongiform encephalopathy caused by abnormal, host-encoded prion proteins that accumulate in the central nervous tissue. CJD occurs in 3 forms: sporadic, familial and acquired (iatrogenic and variant). Six different molecular subtypes (MM1, MM2, MV1, MV2, VV1, and VV2) of sporadic CJD (sCJD) have been identified by genotype at codon 129. The VV1 type represents one of the rarest subtypes. In contrast to the classic sCJD types, these patients are characterized by young age at onset, and slowly progressive dementia. Our case illustrates that sCJD VV1 subtype can have a rapid progression. A 50 year old Caucasian presented with memory loss and personality changes. Magnetic resonance imaging of the brain showed increased signal intensity in the grey mater of cerebral cortex. Brain biopsy confirmed the diagnosis of sCJD VV1 subtype. Currently there is no effective treatment and patient succumbed to death within 3 months of presentation.
Keywords: Creutzfeldt-Jakob disease; Prion diseases; Memory loss
| Introduction | ▴Top |
Prion diseases are a rare group of progressive neurodegenerative disorders with fatal outcome. Creutzfeldt-Jakob disease (CJD) is the most common type of the prion diseases. Its annual incidence worldwide is approximately 1 case per 1 million persons, with the United States reporting less than 300 cases per year [1]. The following case describes a 50-year-old female with rapidly progressive cognitive and motor deficits with temporal lobe biopsy proven diagnosis of a rare subtype of sporadic CJD (sCJD) VV1. This case illustrates that sCJD VV1 subtype can have a rapid progression compared to prior descriptions [2].
| Case Presentation | ▴Top |
A 50-year-old female had been in her usual state of health until approximately 3 months prior to hospital admission. During that time, family members had noticed episodic states of confusion and a "dazed" state of consciousness that waxed and waned. These cognitive symptoms were persistent and worsened during the following 4 weeks, during which the patient also developed a left hand tremor that increased with intentional movement, increasing states of confusion, bradypsychia (slowed mental process), slurred speech and shortened attention span. The patient’s condition worsened by the 6th week of initial presentation with memory loss characterized by failure to recall social security number, internet passwords, and accounting responsibilities, such as balancing check books. During this period the patient’s family members also began to notice an expressive aphasia with slurred speech, broken words and incomplete word pronunciations. By the end of the 8thweek, the patient was unable to complete household chores, had a decreased appetite, and developed left lower extremity weakness with a broad-step gait and bradykinesia (slow motor movement).
By the 12th week from the first onset of symptoms, the woman had an equally degenerative progression of psychomotor and cognitive functions characterized by clumsiness, motor discoordination, lack of hand-eye and alternating hand coordination, left hand tremor and left lower extremity weakness, progressively shortened attention span, loss of short-term memory and poor recall capabilities. At this point the patient was hospitalized complaining of left sided paresis affecting posture, severe dysarthria, bradypsychia, bradycinesia and altered mental status.
The patient’s past medical history was significant for Hodgkin’s lymphoma, for which she received blood transfusion during the course of disease and was treated with MOPP (Mustargen, Oncovin, Procarbazine and Prednisone) and radiation therapy fifteen years ago. There was no history of smoking, alcohol abuse or any illicit drug abuse. Her family history was not significant for any prion disease.
She had traveled to England 20 years back and was a nurse by profession. On neurological examination patient had expressive aphasia, and impaired memory. She had increase tone on the left side with normal reflexes. Cranial nerves were intact. All laboratory results were normal, including blood cell counts, metabolic panel, thyroid functions, antinuclear antibodies, rheumatoid factor, syphilis serology, and HIV test. Cerebrospinal fluid evaluation (CSF) was normal, though it was not evaluated for protein 14-3-3. Electroencephalograph (EEG) demonstrated frontal intermittent rhythmic delta waves suggestive of encephalopathy, an EEG performed two weeks back was normal. Magnetic resonance imaging (MRI) of the brain showed increased signal intensity in the grey mater of cerebral cortex, which was seen best on the diffusion-weighted images. No mass was seen in the deep thalamus, basal ganglia or brainstem (Fig. 1).
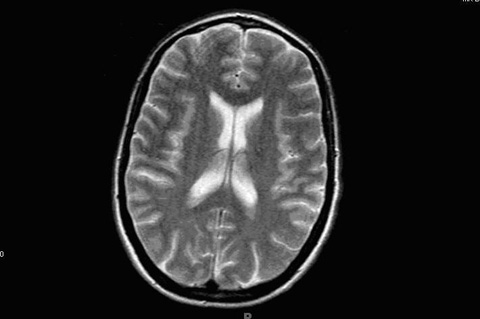 Click for large image | Figure 1. Magnetic resonance imaging (MRI) of the brain showing increased signal intensity in the grey mater of cerebral cortex. |
At this stage of the clinical work-up the following differential diagnoses were considered: MELAS syndrome (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes), mitochondrial encephalopathy, diffuse grey matter disease and prion disease. Upon the recommendation of the various consultants, the decision was made to proceed with a brain wedge biopsy. Due to the unknown etiology and the considered differential diagnosis the specimen obtained from the biopsy was sent to the National Prion Disease Pathology Surveillance Center (NPDPSC). The pathology report from the temporal lobe wedge biopsy suggested a diagnosis of Prion encephalopathy most consistent with CJD. The histopathological and immunohistochemical examination confirmed the diagnosis of prion disease with the characteristics of sCJD, possibly sCJD VV1 subtype (Fig. 2, 3).
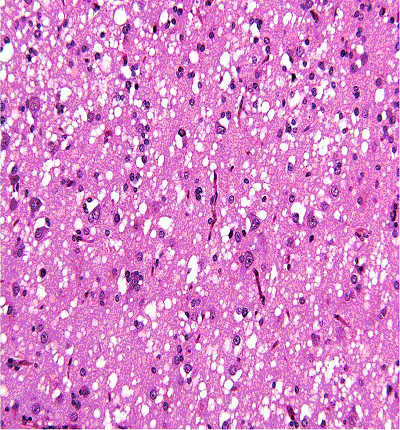 Click for large image | Figure 2. Histopathology (H&E stain) from brain biopsy showing spongiform changes. |
 Click for large image | Figure 3. Immunohistochemical Stain confirming the presence of prion proteins. |
Once the preliminary pathologic report was received, the patient was transferred to an extended-care facility. Over the course of the next few weeks, the patient’s condition continued to deteriorate with worsening memory disturbances, progressive encephalopathy, seizure activity, cerebella ataxia and akinetic mutism. The patient expired within 3 months of presentation.
| Discussion | ▴Top |
Prion diseases are a rare group of progressive neurodegenerative disorders with fatal outcome. In humans it includes CJD, Gerstmann-Straussler-Scheinker disease (GSS), fatal familial insomnia (FFI) and kuru. Each type contains their own unique mechanism of pathogenesis with associated clinical and diagnostic features. The link between each of these prion diseases is the presence of prion protein (PrP) amyloid plaques. These plaques are the result of the conversion of a normal cellular protein PrPc into an abnormal protease resistant isoform [3]. This occurs when the native protein folds in an abnormal fashion, exponentially converting adjacent proteins with a “domino-like” effect, resulting in amyloid plaque formations and structurally damaged neuronal tissue. Neuronal loss, proliferation of glial cells, absence of an inflammatory response and the presence of small vacuoles produce the characteristic spongiform appearance resulting in the common reference of these diseases as the “spongiform encephalopathies”. The prognosis is grim with rapid neurologic decline, ultimately resulting in death within a relatively short period of time.
CJD is the most common of the prion diseases. There are three recognized types: sCJD, familial CJD (fCJD) and acquired {iatrogenic CJD (iCJD) and variant CJD(vCJD)}. Its annual incidence worldwide has steadily hovered around one case per 1 million persons per year, with the United States reporting less than 300 cases per year [1]. Approximately 85% of cases of CJD are sporadic, 5-15% being fCJD and less than 1% iCJD. In 2007, 254 cases were referred to the National Prion Disease Pathology Surveillance Center. Of these, 65 were diagnosed as the sCJD [4].
Researchers have attempted to identify potential risk factors for sCJD. One study in Australia suggested that a history of multiple surgical procedures and residence for more than 10 years on a farm were significant risk factors for sCJD [5]. Another series of studies suggested that a family history of CJD, adjusted odds ratio (OR) = 19.1 or medical history of psychosis (OR = 9.9) were the only significant factors associated with disease [6]. In regards to this case, none of the studies reviewed documented an increased risk of sCJD with receipt of blood products.
Clinical phenotypes of sCJD have been associated with molecular subtypes determined by the PRNP gene codon 129 genotype and the pathologic prion protein (PrPSc) type. The PRNP genotype is homozygous or heterozygous for methionine (M) or valine (V) at codon 129. Using this molecular classification, six clinical phenotypes of sCJD have been described: MM1, MV1, VV2, MV2, MM2 and VV1 [7]. MM1 and MV1 account for about 70% of cases, VV2 for 15%, MV2 for 9%, MM2 for 4% and VV1 for 1%. This case describes a patient with VV1 and represents one of the rarest subtypes [8]. Unlike the typical presentation of the common sCJD types, VV1 patients are characterized by young age of onset, long disease duration and slowly progressive dementia [2]. In contrast to the observations by Meissner et al. [2], this patient presented with rapidly progressive dementia, upper left extremity and postural ataxia with abbreviated disease duration.
This case illustrates that sCJD VV1 subtype can have a rapid progression. The clinical diagnosis can be supported by 14-3-3 in CSF, MRI finding, and confirmed by genetic and molecular analysis. Currently there is no effective treatment for this fatal neurodegenerative disorder.
Primary care physicians are at the forefront of preventing, diagnosing and treating illnesses encountered in the outpatient setting. Referral to subspecialists is appropriate when they are unable to manage certain conditions. The purpose of our training is to not only diagnose and treat disease, but also to address the social, psychological and family dynamics that the illness may cause. The case presented in this paper was written to describe the clinical progression of this rapid evolving encephalopathy, resulting in dementia and gross deterioration of both cognitive and motor functions. The pathology of CJD and its diffuse involvement of the grey matter (as witnessed on MRI) shows how a previously healthy patient develops a constellation of symptoms that progress simultaneously, translating into a substantial and exponential insult to the central nervous system. It is important to keep this “diagnosis of exclusion” within the differential diagnosis when confronted with a case of rapid progressive dementia affecting of both cognitive and motor functions.
A primary care physician encountering such a case in the office setting should consider a prompt work-up and consultation as necessary. These rare diagnoses give them an opportunity to design strategies to council the patient and family members as well as to more efficiently manage end of life issues and care for the patient and their significant others. It is of the utmost importance for the patient’s admitting physician to translate the tremendous and often overwhelming amount of information, in a comprehensible and empathetic fashion: the communication being open to the patient and supportive of the family members concerns.
Conflict of interest
No sources of support and no disclaimers.
| References | ▴Top |
- CJD (Creutzfeldt - Jakob disease, Classic). http://www.cdc.gov/ncidod/dvrd/cjd. CDC 2008.
- Meissner B, Westner IM, Kallenberg K, Krasnianski A, Bartl M, Varges D, Bosenberg C, et al. Sporadic Creutzfeldt-Jakob disease: clinical and diagnostic characteristics of the rare VV1 type. Neurology. 2005;65(10):1544-1550.
pubmed doi - DeArmond SJ, Prusiner SB. Etiology and pathogenesis of prion diseases. Am J Pathol. 1995;146(4):785-811.
pubmed - National Prion Disease Pathology Surveillance Center Cases Examined (NDPDSC). October, 2007.
- Collins S, Law MG, Fletcher A, Boyd A, Kaldor J, Masters CL. Surgical treatment and risk of sporadic Creutzfeldt-Jakob disease: a case-control study. Lancet. 1999;353(9154):693-697.
pubmed doi - Wientjens DP, Davanipour Z, Hofman A, Kondo K, Matthews WB, Will RG, van Duijn CM. Risk factors for Creutzfeldt-Jakob disease: a reanalysis of case-control studies. Neurology. 1996;46(5):1287-1291.
pubmed - Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39(6):767-778.
pubmed doi - Ireland R. Distinguishing VV1 sporadic CJD from variant CJD. Nature Clinical Practice Neurology 2006; 2:64-65.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.