

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 2, Number 2, April 2011, pages 94-96
Autosomal Dominant Polycystic Kidney Disease With Idiopathic Membranous Nephropathy: An Unusual Association?
Antonio Granataa, e, Lidia Puzzob, Fulvio Floccaric, Carmelo Erio Fiored
aDepartment of Nephrology and Dialysis, AOU “Policlinico - Vittorio Emanuele”, Catania, Italy
bDepartment G.F. Ingrassia, Section of Pathology, University of Catania, Catania, Italy
cDepartment of Nephrology and Dialysis, Ospedale San Paolo, Civitavecchia, Italy
dDepartment of Internal Medicine, AOU "Policlinico - Vittorio Emanuele", Catania, Italy
eCorresponding author: Antonio Granata, Via F. Paradiso 78/a, I-95024 Acireale (Catania), Italy
Manuscript accepted for publication February 4, 2011
Short title: ADPKD and Membranous Nephropathy
doi: https://doi.org/10.4021/jmc144w
| Abstract | ▴Top |
Autosomal dominant polycystic kidney disease (ADPKD) is usually characterized by proteinuria less than 1 g/24 hours and only anecdotal cases of associated nephrotic syndrome have been reported. We report another case of ADPKD with nephrotic syndrome, where histological examination showed features of membranous glomerulonephritis. Serologic tests for hepatitis B surface antigen and anti-hepatitis C antibodies were negative. Antinuclear antibody and anti-neutrophil cytoplasmic antibody were negative. Serum complement fraction, C3 and C4, were within normal range. Abdominal ultrasound examination showed both enlarged liver and kidneys, both with multiple cysts of varying sizes. Nevertheless renal parenchyma between cysts was not yet widely interested. Family history revealed ADPKD affecting the mother. Considering nephrotic proteinuria, a percutaneous biopsy was performed, and a diagnosis of idiopathic membranous nephropathy associated with ADPKD was made. Our patient was managed with salt restriction, diuretics and antihypertensive drugs (ACE-I and ARB) and started therapy according to Ponticelli's Protocol. At 12 week follow up proteinuria dropped to less than 1 g/24 hours, with normalization of renal function. Our case confirmed the importance of kidney biopsy even in patients with ADPKD and nephrotic syndrome, in order to demonstrate any coexisting glomerular disease, make an accurate diagnosis and plan appropriate treatment.
Keywords: Nephrotic syndrome; Autosomal dominant polycystic kidney disease; Idiopathic membranous nephropathy; Kidney biopsy
| Introduction | ▴Top |
Autosomal dominant polycystic kidney disease (ADPKD) is usually characterized by proteinuria less than 1 g/24 hours and only anecdotal cases of associated nephrotic syndrome have been reported [1]. Focal segmental glomerulosclerosis, membranous nephropathy, minimal change disease, crescentic glomerulonephritis, IgA nephropathy, mesangioproliferative glomerulonephritis, diabetic glomerulosclerosis and amyloidosis were reported in these cases.
Membranous nephropathy, the most common cause of idiopathic nephrotic syndrome in adults, was reported only in three cases of ADPKD with nephrotic syndrome in literature [1-4]. It is our opinion that we do not perceive correctly epidemiology of glomerulonephritis in these patients, too rarely undergoing renal biopsy. Many nephrologists are in fact probably reluctant to plan a biopsy in polycystic patients, because of perceived increased risk of post-procedural bleeding.
| Case Report | ▴Top |
We here reported a case of ADPKD, affected by nephrotic syndrome, where histological examination showed features of membranous glomerulonephritis.
A 25-year-old man was admitted in our department for swelling of face and feet in the last one month. He denied any history of abdominal pain, fever, dysuria, smoky urine, gross hematuria, sore throat, skin infection, joint pains, skin rash, oral ulcers and using drugs; his blood pressure was 150/100 mmHg. Physical examination was unremarkable, except for bilaterally palpable knobby kidneys and edemas.
Hematological test showed: hemoglobin 141 g/L, total leukocyte count 6880/mm3 (Neutrophils 68%, Lymphocytes 27%), platelet count 3.1 × 105/mm3, blood urea nitrogen 7.2 mmol/L, serum creatinine 179 µmol/L, serum potassium 4.1 mmol/L, serum albumin 28 g/L and serum cholesterol 117.13 mmol/L. Urine analysis showed: 4+ albumin, 10 - 15 leukocytes and 8 - 10 red blood cells/HPF. Urinary protein excretion was 5.27 g/24 hours. Urine culture and throat swab culture were sterile.
Serologic tests for hepatitis B surface antigen and anti-hepatitis C antibodies were negative. Antinuclear antibody (ANA) and anti-neutrophil cytoplasmic antibody (ANCA) were negative. Serum complement fractions, C3 and C4, were within normal range.
Abdominal ultrasound examination showed both enlarged liver and kidneys, both with multiple cysts of varying sizes. Nevertheless renal parenchyma between cysts was not yet widely interested. Family history revealed ADPKD affecting the mother. Based on this finding, the diagnosis of ADPKD was performed.
Considering nephrotic proteinuria, a percutaneous biopsy was performed, with no reported peri-procedural complications. The specimen contained 14 mildly enlarged glomeruli, with rigid appearance of capillary wall, mild thickened basal membrane (PAS +, Congo Red -) with small spike like projections (Silver stain +); direct immunofluorescence on cryostatic slides highlighted granular sub endothelial IgG and C3c (Fig. 1) deposits along glomerular capillary wall, while the other immunoglobulins (IgM, IgA) and complement fractions (C4c, C1q) were normal.
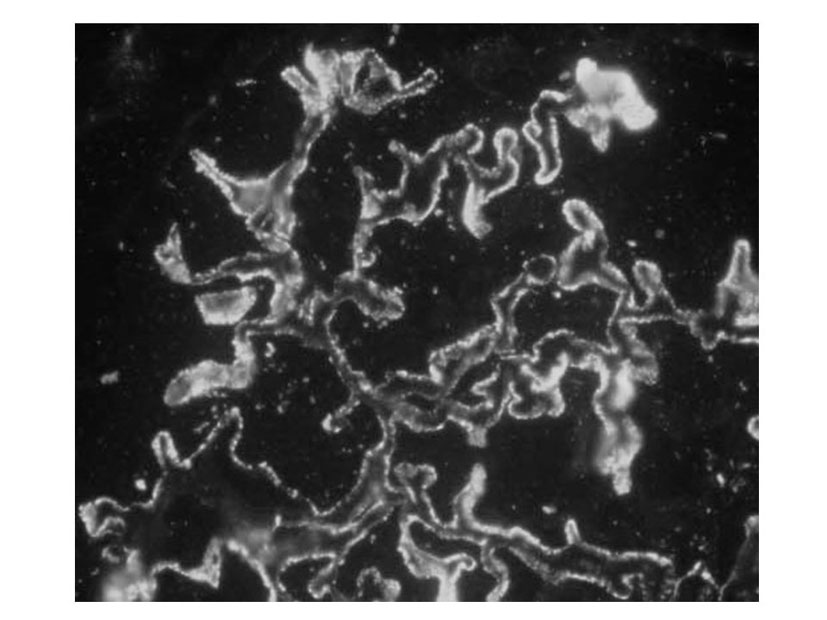 Click for large image | Figure 1. IgG: granular subendothelial deposits along capillary walls. (IF, 40 X) |
Therefore, a diagnosis of idiopathic membranous nephropathy associated with ADPKD was made.
Our patient was managed with salt restriction, diuretics and antihypertensive drugs (ACE-I and ARB) and started therapy according to Ponticelli's Protocol [5]. At 12 week follow up proteinuria dropped to less than 1 g/24 hours, with normalization of renal function.
| Discussion | ▴Top |
This case confirmed the importance of kidney biopsy even in patients with ADPKD and nephrotic syndrome, in order to demonstrate any coexisting glomerular disease, make an accurate diagnosis and plan appropriate treatment.
In our case, among the cysts, kidney parenchyma was preserved, so it was possible to perform a percutaneous needle biopsy instead of an open biopsy. An accurate, preliminary, ultrasonographic and color-Doppler study was performed to plan needle trajectory, in order to avoid vascular structures and cysts. Targeting a cyst-free zone of lower pole allowed us to obtain a procedure totally free of adverse events.
Membranous nephropathy is the most common cause of idiopathic nephrotic syndrome in adults (25%), but it was reported only in few cases of ADPKD with nephrotic syndrome. Is it true that this glomerulonephritis is less frequent in ADPKD patients or this datum is a consequence of nephrologist reluctance to plan biopsies in these subjects?
It is our opinion that if major effort would be spent to make a histological diagnosis, membranous nephropathy will appear as a frequent diagnosis in nephrotic syndrome even in ADPKD population.
| References | ▴Top |
- Contreras G, Mercado A, Pardo V, Vaamonde CA. Nephrotic syndrome in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995;6(5):1354-1359.
pubmed - Saxena S, Hotchandani RK, Bhuyan UN, Agarwal SK, Tiwari SC, Dash SC. Membranous glomerulonephritis associated with autosomal dominant polycystic kidney disease. Nephron 1993;65(2):316-317.
pubmed doi - Shikata K, Makino H, Ota Z. A membranous nephropathy associated with adult polycystic kidney disease. Clin Nephrol 1991;36(5):223-227.
pubmed - Kengne-Wafo S, Massella L, Diomedi-Camassei F, Emma F. Idiopathic membranous nephropathy associated with polycystic kidney disease. Pediatr Nephrol 2010;25(5):961-963.
pubmed doi - Ponticelli C, Passerini P. Management of idiopathic membranous nephropathy. Expert Opin Pharmacother 2010;11(13):2163-2175.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.