








| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 9, Number 7, July 2018, pages 207-210
Occipital Horn Syndrome Case Report: Multidisciplinary Approach of a Rare Entity
Mafalda Mendes-Pintoa, e, Carolina Figueiraa, Ester Pereirab, Margarida Henriquesb, Pedro Louroc, Paula Gouveiaa, Ricardo Veigaa, Paula Garciad
aFunctional Area of Neuroradiology, Medical Imaging Service - Centro Hospitalar e Universitário de Coimbra, Portugal
bPediatrics Service - Hospital de Santo André, Centro Hospitalar de Leiria, Portugal
cGenetics Service - Hospital Pediátrico, Centro Hospitalar e Universitário de Coimbra, Portugal
dDepartment of Metabolic Disorders - Centro de Desenvolvimento da Criança, Hospital Pediátrico, Centro Hospitalar e Universitário de Coimbra, Portugal
eCorresponding Author: Mafalda Mendes-Pinto, Hospitais da Universidade de Coimbra, 3000 - 075 Coimbra, Portugal
Manuscript submitted May 2, 2018, accepted May 25, 2018
Short title: Occipital Horn Syndrome Case Report
doi: https://doi.org/10.14740/jmc3078w
| Abstract | ▴Top |
We report the case of an 8-year-old boy with delayed psychomotor development, dysarthria, choreoathetosis, joint mild hyperlaxity, coarse hair, dry lax skin and facial dimorphisms. Cerebral magnetic resonance revealed diffuse vascular tortuosity, with multiple loops in some arteries of Willis’ circle. Clinical presentation, corroborated by the imagiological findings, raised the suspicion of a disorder of copper metabolism. Serum copper and ceruloplasmin were decreased and lateral skull X-rays demonstrated bilateral occipital exostoses. ATP7A gene sequencing identified a hemizygous variant c.375delA (p.Ala126Glnfs*2), confirming the diagnosis of occipital horn syndrome. Occipital horn syndrome (also called X-linked cutis laxa) is a milder form of Menkes disease. This rare disorder of copper metabolism results from mutations in ATP7A gene encoding a transmembrane copper-transporting enzyme. The broad spectrum of clinical presentation makes the diagnosis challenging. This case illustrates the importance of a multidisciplinary approach, highlighting the role of neuroimaging.
Keywords: Cerebral vascular tortuosity; Bilateral occipital exostoses; Copper metabolism disorders; ATP7A gene; Hemizygous variant c.375delA.
| Introduction | ▴Top |
Occipital horn syndrome, first described in 1975, is a rare entity [1].
This syndrome is an X-linked recessive disorder of copper metabolism. The mutated gene is ATP7A, encoding a copper-transporting ATPase, localized in the trans-Golgi membrane of the cell, and results in abnormal metabolism and distribution of copper [2-4]. It represents a milder form of Menkes disease, characterized by low seric concentrations of normal ATPase and less aggressive phenotype [5]. This syndrome typically causes multisystemic involvement: urinary, gastrointestinal, cardiovascular and/or neurological, as well as connective tissue and musculoskeletal systems [1, 4]. With the cooperation and support of a multidisciplinary team, an early diagnosis is critical, in order to start the imaging follow-up and detect, as earliest as possible, the complications this entity can be associated with.
| Case Report | ▴Top |
We present the case of an 8-year-old Caucasian boy. He was born at term, weighted 3.7 kg (50th percentile) and had an uneventful perinatal period. There was no relevant family medical history. At 2 months of life, he had a right inguinal hernia surgically repaired. Growth parameters were always normal since birth.
At the age of 24 months, his parents were concerned about his language and he was referred to a Pediatrics’ consultation. On follow-up, a psychomotor development delay was diagnosed, with fine motor skills, language and autonomy particularly affected. At clinical examination, notwithstanding a good physical condition, the patient presented with dysarthria and choreoathetosis. He had a coarse hair, dry lax skin, upper joint mild hyperlaxity and facial dimorphisms including long and thin face, high forehead, low-hanging columella, high-arched palate, large ears and darkened tooth enamel.
Brain magnetic resonance imaging (MRI), including angiographic study, demonstrated diffuse tortuosity of arterial vascular structures, including internal carotid and vertebral arteries, as well as anterior, middle and posterior cerebral arteries, presenting some vascular loops (Figs. 1 and 2). Concomitantly, a mild ectasia of transversal sinus (mainly on the right), torcula, straight sinus, ampulla of Galen and terminal portion of the inferior longitudinal sinus with persistence of the falcine sinus was described (Fig. 3). The remaining encephalic study was normal.
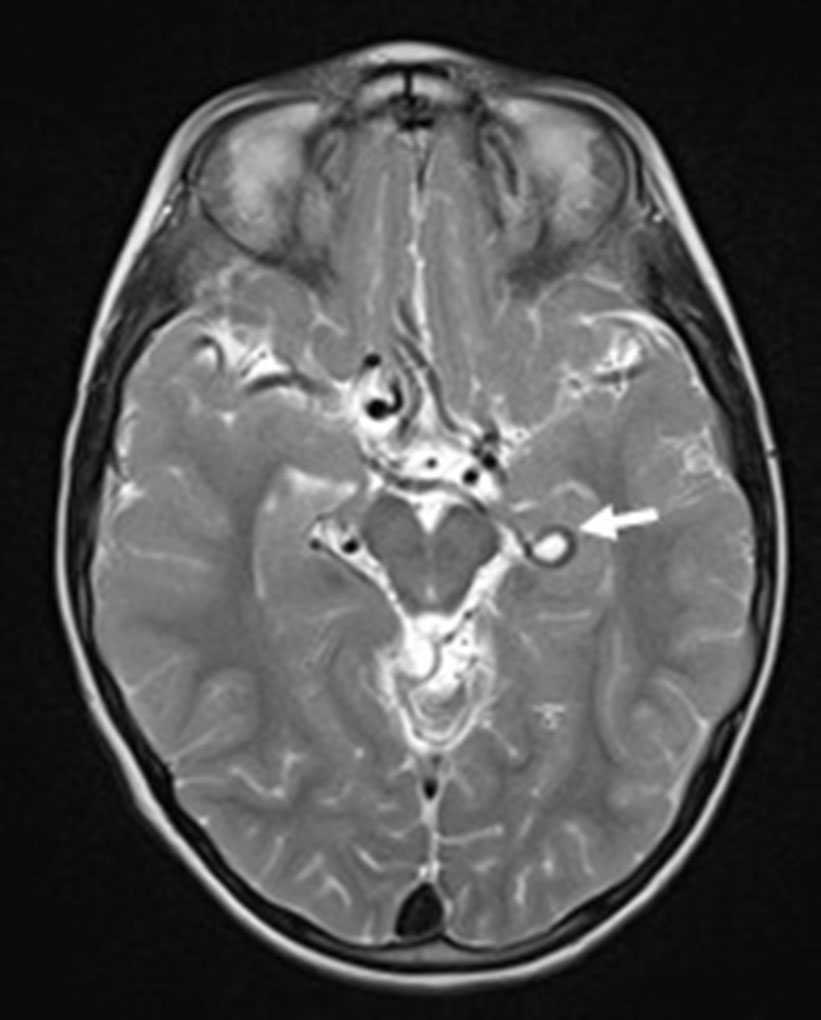 Click for large image | Figure 1. Axial T2-weighted image showing the tortuosity of some Willis’ circle vessels and a looping of the left posterior cerebral artery (arrow). |
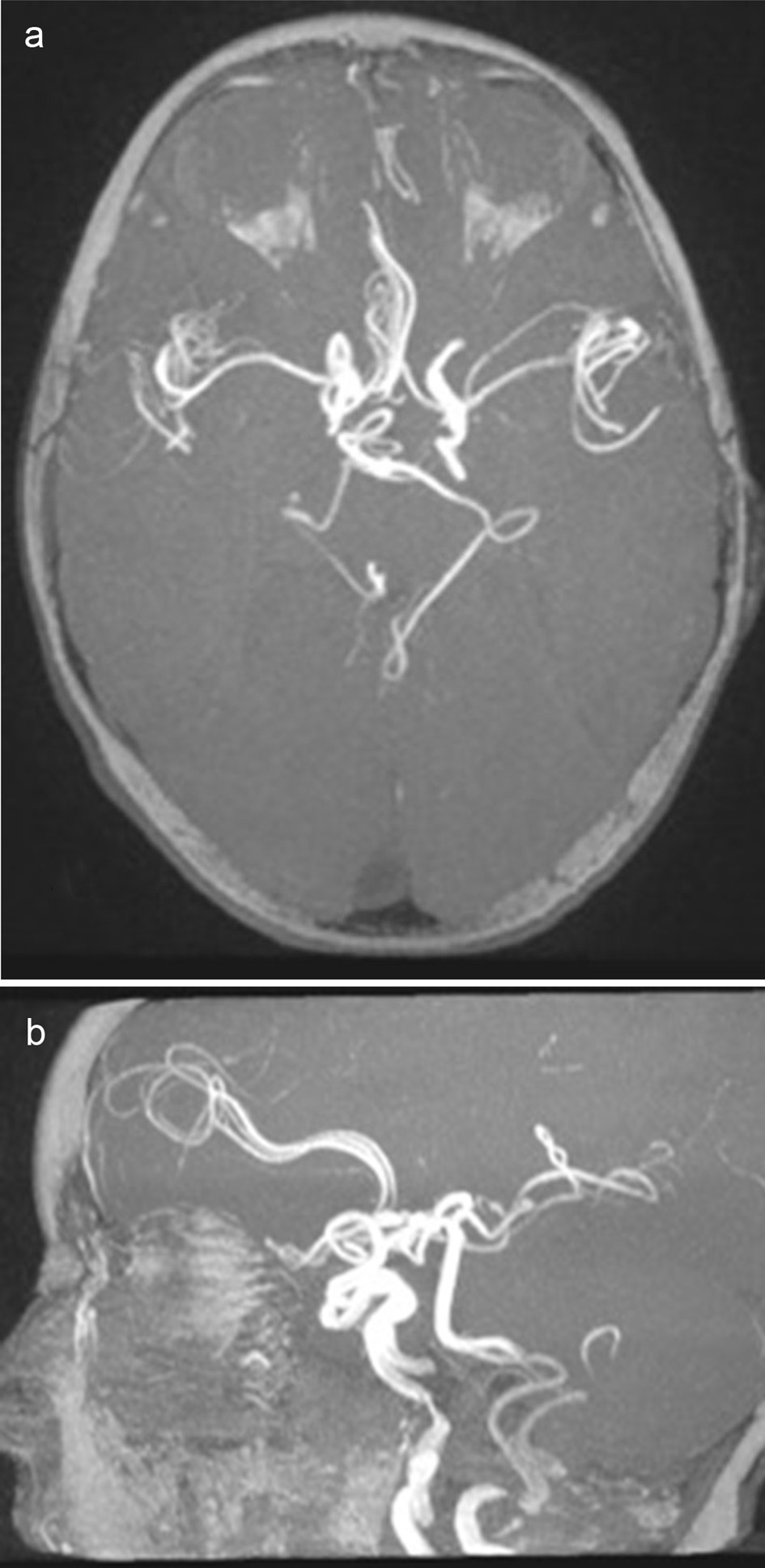 Click for large image | Figure 2. Axial (a) and sagital (b) views of 3D TOF images. Difuse tortuosity of cerebral and cervical arteries. |
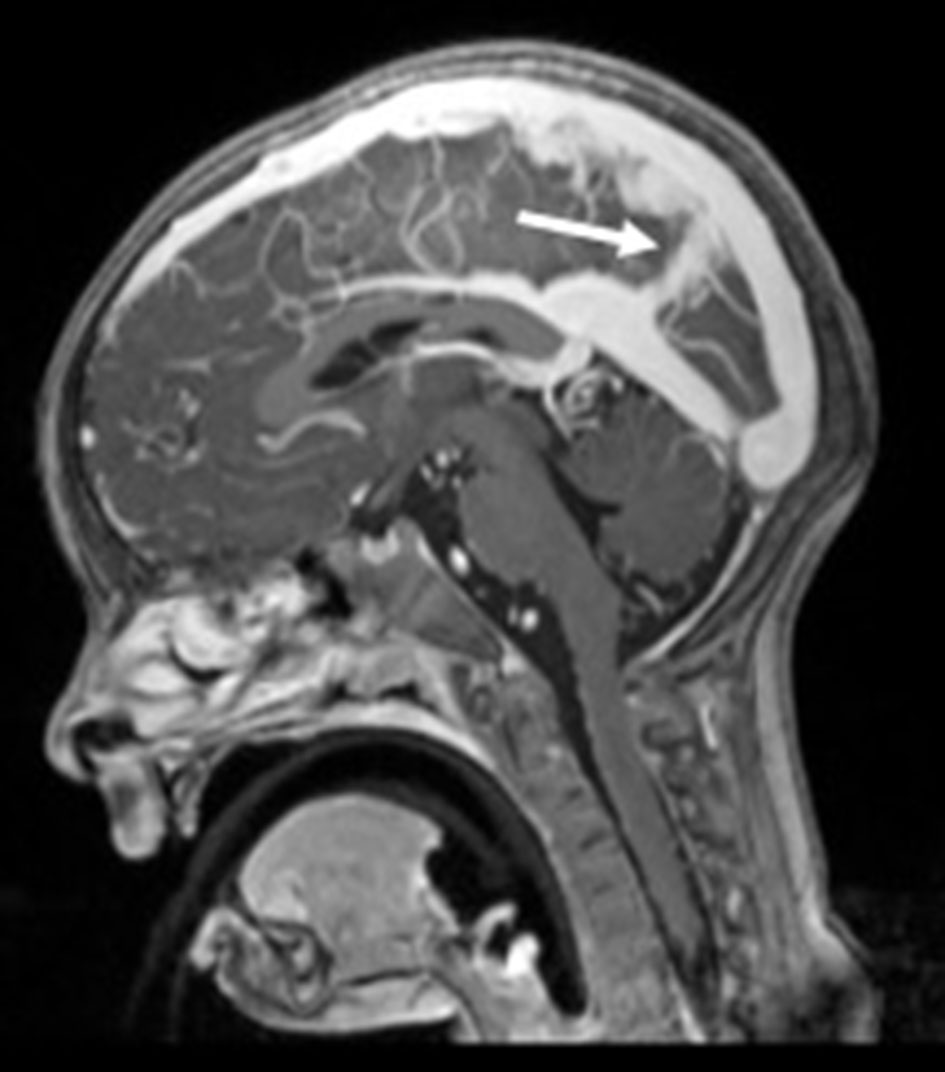 Click for large image | Figure 3. Sagittal T1-MPRAGE-weighted image revealing the persistent falcine sinus (arrow). |
Laboratorial investigations revealed low serum copper (7.6 µmol/L; normal range: 14.1 - 30.0 µmol/L) and low ceruloplasmin (0.16 g/L; normal range: 0.2 - 0.6 g/L).
Thoracic CT angiography, abdominopelvic ultrasound (US) and cardiac study were all normal.
Lateral skull X-ray (Fig. 4) demonstrated bilateral occipital exostoses.
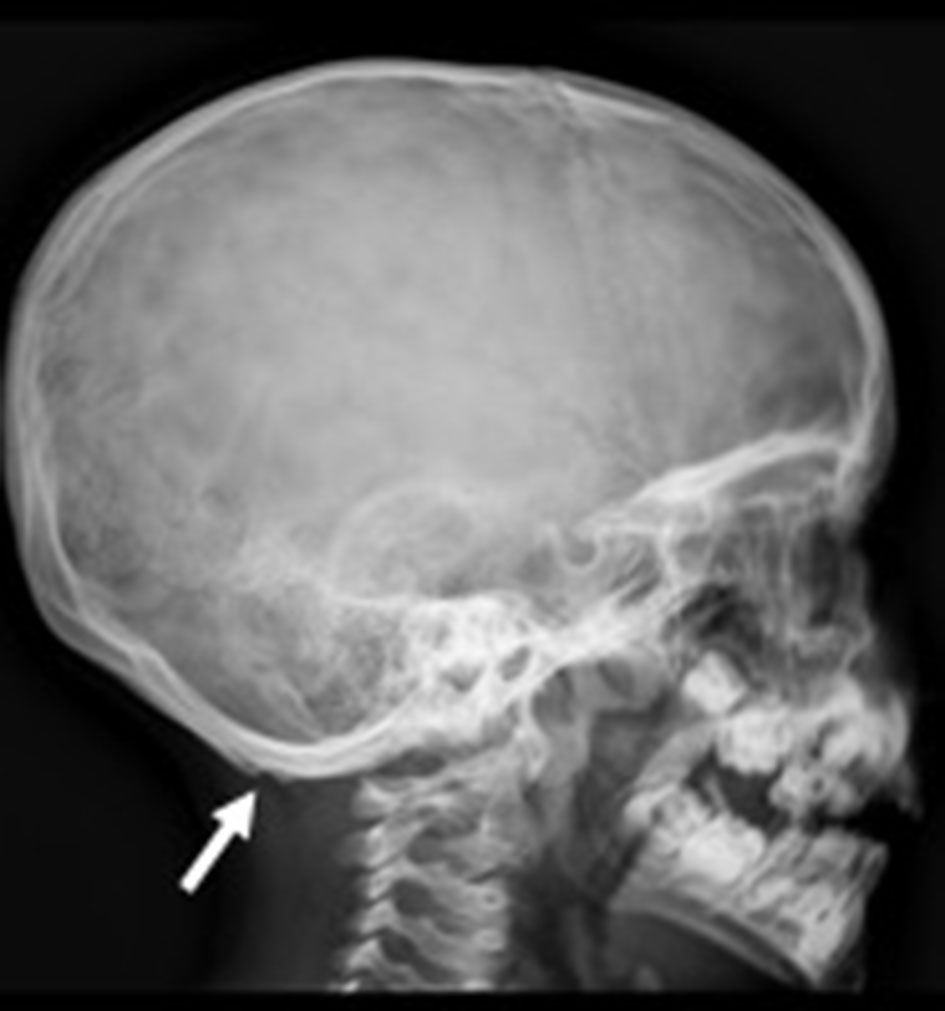 Click for large image | Figure 4. Lateral skull X-ray showing bilateral occipital exostoses (arrow). |
All these clinical, imagiological and laboratorial findings were compatible with occipital horn syndrome and the genetic test was requested. ATP7A gene sequencing identified a hemizygous variant c.375delA (p.Ala126Glnfs*2) that confirmed the diagnosis.
The parents were also studied and the mother was identified as a mutation carrier. They were referred for genetic counselling.
The patient developed a focal epilepsy at 7 years old and began treatment with levetiracetam. Otherwise, he remains stable and attends regular speech therapy, psychomotricity and hydrotherapy sessions. He has bi-annual follow-up at the metabolic out patient clinics of a reference pediatric center.
| Discussion | ▴Top |
Occipital horn syndrome, being an X-linked copper metabolism disorder, typically affects male subjects, while females are usually healthy carriers. Contrary to Menkes disease, whose phenotype is very aggressive and manifests in early stages of life, usually few months after birth, this milder form of disease may be discovered in the infancy or even in the adulthood [2].
As a multisystemic entity, its clinical spectrum includes a wide range of signs and symptoms, with varying severity, from milder to more aggressive forms. These patients usually have facial dimorphisms, as our patient illustrates. Neurological involvement is also documented in the literature, with variable forms of presentation. Usually, they have a moderate-to-severe psychomotor delay, dysarthric speech and seizures [1]. In our patient, it was the psychomotor development delay, including language affection, noticed at 24 months of age, that warned his parents.
Connective tissue manifestations are very typical. Joint and cutaneous hyperlaxity and weakened hair (pili torti) are usually present. Our patient had a dry lax skin, coarse hair and a mild form of upper joint’s hyperlaxity.
Skeletal alterations like the presence of the pathognomonic occipital exostoses (“horns”) that give the name to this entity are typical. They correspond to the tendinous insertions of the trapezius and sternocleidomastoid muscles in the occipital bone, probably resulting from an inflammatory reaction to chronic injury [6].
Our patient’s lateral skull X-ray confirmed the presence of bilateral occipital horns. Other skeletal abnormalities such as pectum excavatum or carinatum, cervical and lumbar cifosis, osteopenia, among others, were absent [7].
Other systems, like the urinary and the gastrointestinal, may be involved, with variable levels of severity: inguinal herniation, bladder diverticulum with obstructive uropathy, as well as chronic diarrhea [7]. Our patient had a right inguinal hernia surgically repaired. Other characteristic aspect is the vascular involvement, namely, with tortuous vessels, due to the fragility of the arteries’ walls with thinning and fragmentation of the internal elastic lamina and defective elastin fibers [1].
In this case report, we underline the relevance of this finding, since the discovery of cerebral and cervical tortuous arteries in the MRI study should trigger diagnosis suspicion.
However, vascular tortuosity is a non-specific finding as it can be present in some other entities, in a more or less generalized form. Some examples are the arterial tortuosity syndrome, EFEMP2-related cutis laxa (autosomal recessive cutis laxa type 1B), or Loeys-Dietz syndrome, among others [8].
In our case report, a copper’s metabolism disease was considered by clinical presentation and corroborated by the imagiological findings. Cerebral and cervical vessels were the only ones involved.
Laboratorial studies revealing low copper and ceruloplasmin levels in the blood and high copper levels in cultured fibroblasts are suggestive of occipital horn syndrome [2]. However, genetics has nowadays the main role in this diagnostic process, with the identification of the ATP7A gene mutations enabling the definitive diagnosis [3].
In the reported case, ATP7A gene sequencing identified a hemizygous variant c.375delA (p.Ala126Glnfs*2). This was considered a unique finding, whose predicted outcome was a complete loss of function. Nevertheless, our patient’s milder phenotype probably results in inefficient translational re-initiation, enabling some functional copper transport.
Conclusions
Occipital horn syndrome may be considered a “difficult-to-diagnosis” entity, not only because of its rarity, but also because it has a variable, heterogeneous and sometimes unspecific clinical involvement, whose severity levels can also be irregularly defined. A correct and early diagnosis is important in order to start an appropriate follow-up, preventing severe complications.
With this case report, we highlight the importance of a multidisciplinary team, where clinics and diagnostic studies complement each other, making the diagnostic workup easier and faster.
Conflict of Interest
None.
Grant Support
None.
| References | ▴Top |
- Palmer CA, Percy AK. Neuropathology of occipital horn syndrome. J Child Neurol. 2001;16(10):764-766.
doi pubmed - Bazzocchi A, Femia R, Feraco P, Battista G, Canini R, Guglielmi G. Occipital horn syndrome in a woman: skeletal radiological findings. Skeletal Radiol. 2011;40(11):1491-1494.
doi pubmed - Tumer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013;34(3):417-429.
doi pubmed - Yasmeen S, Lund K, De Paepe A, De Bie S, Heiberg A, Silva J, Martins M, et al. Occipital horn syndrome and classical Menkes Syndrome caused by deep intronic mutations, leading to the activation of ATP7A pseudo-exon. Eur J Hum Genet. 2014;22(4):517-521.
doi pubmed - Dozza ALCB, Fernandes GD, Yuen CT, Santanna BA, Souza KS, Araujo APQC. Doenca de Menkes: Relato de Caso. Rev Bras Neurol. 2009;45(4):43-47.
- Gerard-Blanluet M, Birk-Moller L, Caubel I, Gelot A, Billette de Villemeur T, Horn N. Early development of occipital horn in a classical Menkes patient. Am J Med Genet A. 2004;1:2:11-13.
doi - Lobo AL et al. Sindrome da Cutis Laxa Ligada ao X: Caso Clinico. Acta Pediatr. Port. 2003;34:295-297.
- Callewaert B, De Paepe A, Coucke P. Arterial Tortuosity Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, eds. GeneReviews. University of Washington, Seattle WA, 1993.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.