








| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 9, Number 2, February 2018, pages 73-75
Woodhouse-Sakati Syndrome With Psychosis and Basal Ganglia Calcification: A Case Report
Trad Abdulaziz Alasiria, c, Adel Ahmed Alshehria, Ahmed Saad Alzahranib
aCollege of Medicine, Umm Al-Qura University, Mecca, KSA
bNeuroscience Center, King Abdullah Medical City, Mecca, KSA
cCorresponding Author: Trad Abdulaziz Alasiri, College of Medicine, Umm Al-Qura University, Mecca, KSA
Manuscript submitted December 24, 2017, accepted January 15, 2018
Short title: Woodhouse-Sakati Syndrome Case Report
doi: https://doi.org/10.14740/jmc2996w
| Abstract | ▴Top |
Woodhouse Sakati syndrome is a rare autosomal recessive multisystem disorder characterized by neuropsychological, endocrinal, and ectodermal symptoms. We report a 52-year-old Saudi female with psychosis, extrapyramidal signs, sensorineural hearing loss, diabetes, hypothyroidism, amenorrhea, hypergonadotropic hypogonadism, absence of secondary sexual characters, and alopecia totalis. Her brain computed tomography (CT) showed bilateral basal ganglia calcification and her genetic testing confirmed homozygous single nucleotide deletion (C.436delC) of frameshift mutation (p.A147HfsX9) in exon 4 of the gene DCAF17 gene. Woodhouse Sakati syndrome seems to be more prevalent, and probably underdiagnosed, in the Middle East and Arab countries, and should be considered in patients with a combination of neurological and endocrinal dysfunction. To our knowledge, this is the first case reported to have psychotic symptoms.
Keywords: Woodhouse Sakati syndrome; Case report; Saudi Arabia; Psychosis; Basal ganglia calcification
| Introduction | ▴Top |
Woodhouse Sakati syndrome is a rare autosomal recessive disorder that affects multiple systems particularly the endocrinal and the neurological systems [1]. Its prevalence is unknown, probably < 1/1,000,000 [2]. So far, only few families were reported in the medical literature, especially in the Middle East region [1]. Woodhouse Sakati syndrome is characterized by various clinical symptoms including endocrinal dysfunction (especially diabetes mellitus, hypothyroidism, and hypogonadism), ectodermal manifestations (such as alopecia, wrinkled facial skin, and anodontia), and neurological impairment (including extrapyramidal symptoms, intellectual dysfunction, psychosis, and sensory neural hearing loss) [3]. Woodhouse Sakati syndrome is important to be considered in patients experiencing any combination of these aforementioned symptoms. Because of the heterogenicity of the disease phenotype, patients may present differently as regards the symptoms, the age at onset, the severity of manifestations and the disease course [3].
We report here another Saudi female with a history, clinical examination, and investigations suggesting the diagnosis of Woodhouse Sakati syndrome.
| Case Report | ▴Top |
We report a 52-year-old lady who was born to consanguineous parents (first degree parenteral cousins). She had normal developmental growth but her scholastic performance was poor due to perceived intellectual difficulties. Soon after her first year of school, she was dropped out. At age of puberty, the patient did not develop secondary sexual characteristics and she had primary amenorrhea.
At the age of 24 years, deafness evolved insidiously and the patient’s tympanometry revealed type C pattern in the right ear and type A pattern in the left ear with loss of acoustic reflexes at all frequencies. Nine years later, the patient was diagnosed with diabetes mellitus that presented with disturbed consciousness. At this time, full hormonal profile was recommended that confirmed diabetes mellitus, gonadal dysfunction, and revealed hypothyroidism. At the age of 46 years, she began to complain of insidious progressive visual dysfunction. After seeking medical advice, her fundus examination revealed bilateral optic atrophy.
During the fourth decade of life, she experienced psychotic manifestations in the form of auditory and visual hallucinations, delusion of persecution and disorganized speech. Auditory hallucinations were non-commanding and non-commenting in nature but mainly unrecognized voices or animal voices. Visual hallucinations were of animals, known and unknown people to the patient, but none were fearful or threatening. However, her delusion of persecution was occasionally tense and fearful to the patient in which she believed that her close brother as well as other unfamiliar persons was trying to kill her. She was diagnosed as schizophrenia and received olanzapine initially. The dose of olanzapine was optimized to 40 mg per day over approximately 7 years but with a remitting course and minimal improvement. Moreover, she was on other different medications as aripiprazole and lamotrigine. However, more recently and because of the minimal improvement in her psychotic symptoms and functioning, she was switched to clozapine as a last resort which was titrated gradually to 200 mg twice daily. Her delusions and hallucinations have been mostly controlled and her fears have been subsided.
Patient physical examination revealed alopecia totalis, loss of eyebrow hair, and eyelashes. Her secondary sexual characteristics were absent. She had no axillary or pubic hair, and breast hypoplasia. Her neurological examination showed resting tremor and peripheral neuropathy (mostly due to diabetes).
As regards the laboratory investigations, the patient’s hormonal profile revealed high TSH (5 mIU/L, normal 0.4 - 4.0 mIU/L), low FT4 (7, normal 9.0 - 25.0), low FT3 (2.4, normal 3.5 - 7.8), high luteinizing hormone level (13 mIU/L, normal range 1.8 - 11.78 mIU/L), high follicle-stimulating hormone level (40 mIU/L, normal range 3.03 - 8.08 mIU/L), high Hb A1c (10.5%, normal 4% - 5.6%), normal ACTH (12 pcg/mL, 9 - 52 pcg/ml), normal cortisol (6 µg/dL, normal 10 - 20 µg/dL), normal growth hormone level (0.337 ng/mL, normal < 10 ng/mL), and normal prolactin (6.7 ng/mL, normal 2 - 29 ng/mL). These laboratory data are suggestive of diabetes mellitus, hypothyroidism, and hypogonadotropic hypogonadism.
Computed tomography (CT) of the brain (Fig. 1) showed bilateral basal ganglia calcification with diffuse cerebral atrophy. Her electroencephalogram (EEG) showed slow background activity. Her ophthalmologic examination revealed bilateral optic atrophy, and her hearing assessment ensured loss of acoustic reflexes bilaterally. Type A and type C tympanometry were demonstrated in left and right ears, respectively. Nerve conduction velocity study revealed diabetic sensorimotor polyneuropathy.
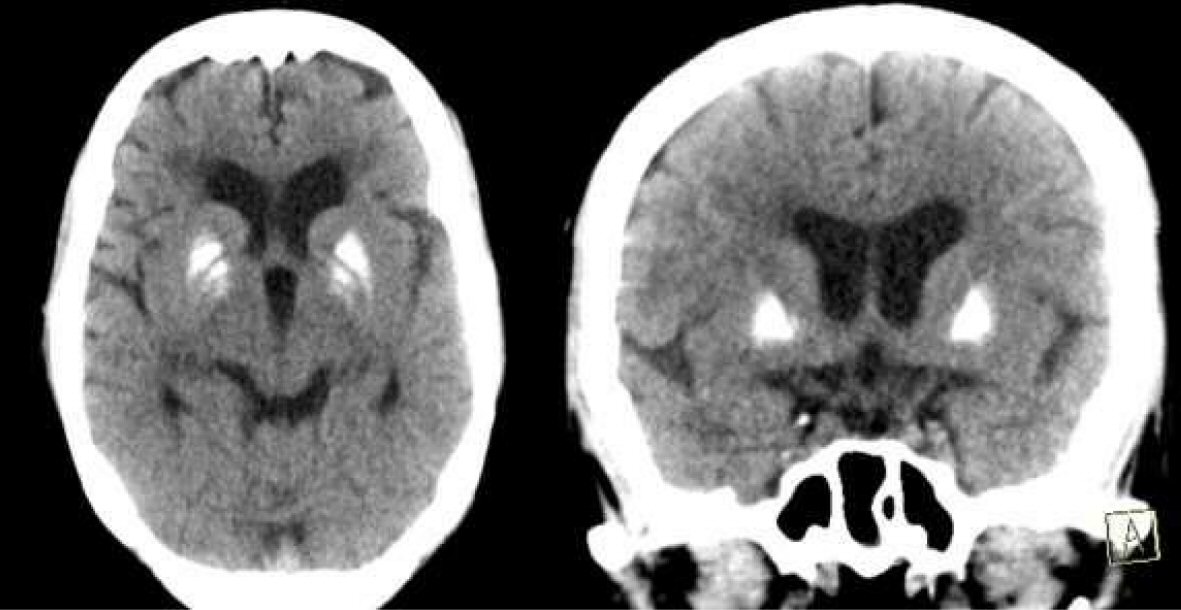 Click for large image | Figure 1. CT brain showing bilateral basal ganglia calcification with involutional brain changes. |
The patient was tested for DCAF17 gene mutations by polymerase chain reaction (PCR) and direct consequence. DCAF17 gene maps to chromosome 2q31.1. The gene comprises 14 exons and encodes 520 amino acid proteins with “ATG”. Genomic DNA was used from this patient to amplify the 14 coding exons and the corresponding flanking sequences of the DCAF17 gene. The results of her genetic testing showed that she had homozygous disease-causing single nucleotide deletion (C.436delC) of frameshift mutation (p.A147HfsX9) in exon 4 of the gene DCAF17 gene.
| Discussion | ▴Top |
Woodhouse Sakati disease is a very rare multisystem degenerative disorder that typically affects the neurological and endocrinological systems. It is considered one of the neurodegeneration with brain iron accumulation disorders [4]. Our patient presented with classical features of the disease including neurological, endocrinal, and ectodermal symptoms [3]. She had primary amenorrhea, with high FSH and LH level, suggesting hypergonadotropic hypogonadism. This type of hypogonadism was reported in some previous case reports [5]. She also developed insulin dependent diabetes mellitus at her third decade which was a typical presentation in previously reported patients [1, 6]. Her thyroid profile revealed that she had thyroid dysfunction of peripheral origin, which is common in 30% of cases of Woodhouse syndrome [3, 6]. Prolactin and cortisol levels were within normal range. As regards the ectodermal manifestations, the patient developed alopecia totalis which was reported in almost all cases with Woodhouse Sakati syndrome [3]. Additional symptoms included sensorineural hearing loss, visual impairment, extrapyramidal signs, delusion of persecution, visual and auditory hallucinations, and intellectual dysfunction.
Neurophysiological testing of this patient revealed demyelinating sensory motor polyneuropathy, which probably attributed to diabetes mellitus. Schneider and Bhatia reported similar peripheral neuropathy in their reported family [7]. EEG revealed generalized slowing, but our patient did not have any seizures. As regards brain imaging, the CT brain of our patient showed bilateral basal ganglia calcification which was not reported before. The calcification seems to be idiopathic because her calcium profile was normal. Previous reported studies reported variable brain imaging abnormalities; some patients had variable abnormalities at deep white matter [6, 7], whereas others had iron deposition in globus pallidus [8, 9].
Being born to consanguineous parents and having a brother with the same condition suggested an autosomal recessive inheritance. Because defects in DCAF17 (OMM312515) are responsible for causing the disease, the patient was tested for mutations of this gene, and her genetic testing revealed homozygous disease-causing single nucleotide deletion (C.436delC) of frameshift mutation (p.A147HfsX9) in exon 4 of the gene DCAF17 gene. This mutation has been previously reported in patients with Woodhouse Sakati syndromes [9-11].
In spite of being rare, Woodhouse syndrome should be considered in patients presenting with combinations of neuropsychological, endocrinal, and ectodermal manifestations, especially in the Middle East and Arab world. To date, only few dozens of families were reported to have the disease, especially in Saudi Arabia [5, 6, 11], Pakistan [12], Tunisia [9], Turkey [13], Croatia [14], India [15], and Italy [16]. The cases reported have various clinical, phenotypic, and genotypic patterns. Thus, it is of importance to report and study the clinical presentation of different cases with Woodhouse Sakati syndrome to have a better understanding of the disorder.
Acknowledgments
The authors would like to thank the patient, and her family for going through the trouble in the continuous ongoing testing and for letting us share this case with the medical community.
Conflict of Interest
All the authors have reported no conflict of interest.
Grant Support
None.
| References | ▴Top |
- Agopiantz M, Corbonnois P, Sorlin A, Bonnet C, Klein M, Hubert N, Pascal-Vigneron V, et al. Endocrine disorders in Woodhouse-Sakati syndrome: a systematic review of the literature. J Endocrinol Invest. 2014;37(1):1-7.
doi pubmed - Orphanet: Woodhouse Sakati syndrome. [Online]. Available: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=3464. [Accessed: 13-Nov-2017].
- Bohlega SA, Alkuraya FS. Woodhouse-Sakati Syndrome. University of Washington, Seattle; 1993.
- Salomao RP, Pedroso JL, Gama MT, Dutra LA, Maciel RH, Godeiro-Junior C, Chien HF, et al. A diagnostic approach for neurodegeneration with brain iron accumulation: clinical features, genetics and brain imaging. Arq Neuropsiquiatr. 2016;74(7):587-596.
doi pubmed - Woodhouse NJ, Sakati NA. A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J Med Genet. 1983;20(3):216-219.
doi - Al-Semari A, Bohlega S. Autosomal-recessive syndrome with alopecia, hypogonadism, progressive extra-pyramidal disorder, white matter disease, sensory neural deafness, diabetes mellitus, and low IGF1. Am J Med Genet A. 2007;143A(2):149-160.
doi pubmed - Schneider SA, Bhatia KP. Dystonia in the Woodhouse Sakati syndrome: A new family and literature review. Mov Disord. 2008;23(4):592-596.
doi pubmed - Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep. 2011;11(3):254-261.
doi pubmed - Hdiji O, Turki E, Bouzidi N, Bouchhima I, Damak M, Bohlega S, Mhiri C. Woodhouse-Sakati syndrome: report of the first tunisian family with the C2orf37 gene mutation. J Mov Disord. 2016;9(2):120-123.
doi pubmed - Alazami AM, Al-Saif A, Al-Semari A, Bohlega S, Zlitni S, Alzahrani F, Bavi P, et al. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am J Hum Genet. 2008;83(6):684-691.
doi pubmed - Ben-Omran T, Ali R, Almureikhi M, Alameer S, Al-Saffar M, Walsh CA, Felie JM, et al. Phenotypic heterogeneity in Woodhouse-Sakati syndrome: two new families with a mutation in the C2orf37 gene. Am J Med Genet A. 2011;155A(11):2647-2653.
doi pubmed - Habib R, Basit S, Khan S, Khan MN, Ahmad W. A novel splice site mutation in gene C2orf37 underlying Woodhouse-Sakati syndrome (WSS) in a consanguineous family of Pakistani origin. Gene. 2011;490(1-2):26-31.
doi pubmed - Gul D, Ozata M, Mergen H, Odabasi Z, Mergen M. Woodhouse and Sakati syndrome (MIM 241080): report of a new patient. Clin Dysmorphol. 2000;9(2):123-125.
doi pubmed - Medica I, Sepcic J, Peterlin B. Woodhouse-Sakati syndrome: case report and symptoms review. Genet Couns. 2007;18(2):227-231.
pubmed - Koshy G, Danda S, Thomas N, Mathews V, Viswanathan V. Three siblings with Woodhouse-Sakati syndrome in an Indian family. Clin Dysmorphol. 2008;17(1):57-60.
doi pubmed - Steindl K, Alazami AM, Bhatia KP, Wuerfel JT, Petersen D, Cartolari R, Neri G, et al. A novel C2orf37 mutation causes the first Italian cases of Woodhouse Sakati syndrome. Clin Genet. 2010;78(6):594-597.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.