*Average article statistics from the last 12 months data
COVID-19 RESEARCH
The COVID-19 outbreak presents the unprecedented challenge for world public and medical practitioners and health care providers, the post COVID-19 condition (or long COVID) includes long term symptoms which may persist for months or years after SARS-CoV-2 infection. We will consider submissions related to all aspects of COVID-19 and Long COVID, and process the manuscripts in priority.
Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access
Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc
Journal website http://www.journalmc.org
Case Report
Volume 8, Number 7, July 2017, pages 203-206
Anesthetic Management of a 13-Year-Old Adolescent With Mucolipidosis Type II for Total Hip Arthroplasty
Mumin Hakima, c, Hina Waliaa, Senthil G. Krishnaa, b, Joseph D. Tobiasa, b
aDepartment of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, Columbus, OH, USA bDepartment of Anesthesiology, The Ohio State University, Columbus, OH, USA cCorresponding Author: Mumin Hakim, Department of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, 700 Children’s Drive, Columbus, OH 43205, USA
Manuscript submitted April 21, 2017, accepted May 12, 2017 Short title: Anesthesia and Mucolipidosis Type II doi: https://doi.org/10.14740/jmc2834w
Mucolipidosis II (ML II) or inclusion cell disease is a rare lysosomal storage disorder, inherited as an autosomal recessive trait. Deficiency of the lysosomal transport enzyme, N-acetylglucosamine-1-phosphotransferease, results in the intracellular accumulation of macromolecules (mucopolysaccharides and mucolipids) in the lysosome which leads to cellular dysfunction and a multi-system disorder. Manifestations are present at birth including muscle hypotonia, a weak cry, and failure to thrive. Additional physical signs include hip dislocation, inguinal hernia, hepatomegaly, limitation of joint movement, and cutaneous changes. Coarse facial features and skeletal abnormalities become more conspicuous with time. A rapidly progressive psychomotor deterioration, developmental delay and growth failure are often noted with a limited life span of 10 years. Due to the aforementioned physical abnormalities, surgical and anesthetic care may be required. We present a 13-year-old girl with ML II who required anesthetic care for a total hip arthroplasty due to chronic right hip dislocation. Previous reports of anesthetic care for these patients are reviewed, end-organ involvement is discussed, and options for anesthetic care are presented.
Keywords: Hip dislocation; Mucolipidosis II; I-cell disease
Mucolipidosis II (ML II) or inclusion cell disease is a rare, progressively debilitating, and fatal lysosomal storage disorder, which was first described by Leroy and DeMars in 1967 [1]. The exact prevalence of ML II is unknown with an estimated incidence of 1 in 100,000 - 400,000 individuals worldwide. Due to its rarity, clinical history and management considerations for these children have not been established [2, 3]. Patients have a limited life expectancy of less than 10 years in most cases with death secondary to cardiopulmonary complications including recurrent upper respiratory tract infections, bronchopneumonia, and heart failure [4, 5].
ML II is an autosomal recessive metabolic storage disorder, resulting from a deficiency of the lysosomal transport enzyme, N-acetylglucosamine-1-phosphotransferease, which is one of the two enzymes required for the biosynthesis of the mannose 6-phosphate (M6P) recognition marker [2]. M6P is a common marker which facilitates the transport of enzymes into the lysosomal compartment of all cells. Without this marker, lysosomal endocytosis is impaired secondary to an improper intracellular trafficking signal. This results in the diversion of lysosomal enzymes into the extra-cellular space, thereby causing an elevation of lysosomal enzymes in plasma [6, 7]. The lack of lysosomal enzymes results in the accumulation of macromolecules (mucopolysaccharides and mucolipids) in the lysosome resulting in an abnormal architecture including coarse cytoplasmic granular inclusions in cultured skin fibroblasts giving rise to the name, inclusion cell or “I-cell” disease [8].
Although initially there were four types of ML (I, II, III and IV), the biochemical causes of ML II and III were further identified as different from the other lysosomal storage diseases, the mucopolysaccharidoses (MPS) such as Hurler’s and Hunter’s disease. In the latter, there is only one lysosomal enzyme that is deficient in each disease because of a mutation in the responsible gene. Alpha-L-iduronidase is deficient in MPS I while in ML II and III, alpha-L-iduronidase is one of the many enzymes lacking in lysosomes as the defect lies in the targeting signal which results in the transport of several different enzymes into the lysosome. This explains why I-cell disease has some of the same clinical features as MPS I as well as other clinical manifestations. We present a 13-year-old adolescent with ML II who required anesthetic care for a total hip arthroplasty due to chronic right hip dislocation. Previous reports of anesthetic care for these patients are reviewed, the end-organ involvement is discussed, and options for anesthetic care are presented.
Institutional Review Board approval is not required at Nationwide Children’s Hospital (Columbus, OH) for publication of one case report. The patient was a 13-year-old, 22.4 kg girl with ML type II. She presented for total hip arthroplasty due to chronic right hip dislocation. Her past medical history included aortic and mitral regurgitation, cardiomegaly, developmental delay, congenital hip dysplasia, kyphoscoliosis, gastroesophageal reflux, and reactive airway disease. Her past surgical history included umbilical hernia repair, adenotonsillectomy, and multiple previous orthopedic procedures. She had a known allergy to morphine and has previously reacted in the form of hives. Her long-term outpatient medication included diazepam 2 mg PO QID, methadone 5 mg PO TID, oxycodone 5 mg PO TID, magnesium 250 mg PO OD, docusate sodium 50 mg PO OD, aspirin 81 mg PO OD and duloxetine 20 mg PO OD. Preoperative laboratory evaluations including electrolytes and renal function, coagulation function, blood glucose, and hepatic function were normal. On physical examination, craniofacial structures were asymmetric with a small mouth and a limited thyromental distance (less than three finger breaths). A recent echocardiogram showed mild aortic regurgitation, moderate mitral regurgitation, and mild mitral valve prolapse. Overall myocardial function was normal. She was held nil per os for 8 h for solids and 3 h for liquids prior to surgery and was transported to the operating room where routine American Society of Anesthesiologists’ monitors were applied. A pre-existing peripheral intravenous catheter was in place. Anesthesia was included with midazolam (0.1 mg/kg), fentanyl (1 µg/kg), and propofol (3 mg/kg). Bag-valve-mask ventilation was non-problematic and a 2.5 laryngeal mask airway (LMA) was placed atraumatically. Anesthesia was maintained with sevoflurane in air/oxygen. Skeletal muscle relaxation was achieved with a single dose of rocuronium (0.5 mg/kg) to aid the closed reduction maneuver. The reduction was confirmed on intraoperative fluoroscopy. Following the completion of the procedure, sugammadex (2 mg/kg) was administered to reverse neuromuscular blockade. Following emergence from anesthesia, the LMA was removed and the patient was transported to post-anesthesia care unit in stable condition. The remainder of the postoperative course was uneventful.
ML II is a rare lysosomal storage disorder, inherited as an autosomal recessive trait. Severe developmental delay and failure to thrive are the most common presenting symptoms [2, 9]. At birth, these patients are typically underweight with generalized hypotonia and a weak cry. Failure to thrive is common with growth arrest during the second year of life. Infants develop distinctive facies with coarse facial features including a high, narrow forehead, puffy eyelids, epicanthal folds, flat nasal bridge, anteverted nares, long philtrum, prominent gingival hyperplasia and macroglossia. As the disorder results in the absence of normal lysosomal enzymes, it shares many clinical features with the MPS including cardiac, pulmonary, and airway involvement. Additional clinical manifestations include mental and physical retardation with typical orofacial feature. The lower part of the face usually has a “fishlike” profile due to enlargement of the gingivae and alveolar process. Progressive gingival hypertrophy gives rise to an open bite. A hoarse voice is noted due to infiltration of the glottis structures and stiffening of vocal cords. Hearing loss can result due to recurrent ear infections and infiltration of middle ear structures. Musculoskeletal features include contractures affecting mobility, kyphosis, short hands, unusually shaped long bones and dislocated hips. Other features include umbilical and/or inguinal hernia, hepatomegaly, heart valve anomalies including aortic and mitral insufficiency, and corneal opacities [10, 11]. A constellation of radiographic abnormalities which consists of abnormally shaped vertebrae and ribs, enlarged skull, spatulate ribs, hypoplastic epiphyses, thickened diaphyses and bullet-shaped metacarpals, known as dysostosis multiplex is exhibited in ML II patients [10, 11].
In addition to the phenotypic manifestations, confirmatory diagnostic tests include marked elevations of lysosomal enzymes in the plasma with the presence of large lysosomal inclusions in peripheral lymphocytes [2, 7]. The only therapeutic approach currently available for ML II is bone marrow transplantation which supplies a source of structurally normal lysosomal enzymes [12].
Major perioperative concerns include involvement of the airway, respiratory and cardiovascular systems. As with the more common MPS, airway management and endotracheal intubation may be problematic in patients with ML II. Airway involvement includes infiltration and distortion of upper and lower airway structures, abnormal physical features (microstomia, micrognathia, macroglossia, and malocclusion), and limited mouth opening. These issues may be further magnified by musculoskeletal malformations with limited neck movement. Mucopolysaccharide deposits in the supraglottis and anterolateral displacement of the larynx have led to difficulty with mask ventilation and other attempts at direct laryngoscopy, flexible endoscopy, and indirect laryngoscopy including indirect video laryngoscopy using the Glidescope® [13-16]. A previous study which looked at the practical challenges faced in securing airways revealed that the ML II group of children required an average of 3.6 attempts to achieve endotracheal intubation compared to 1.2 in the control group [13, 17]. The appropriate equipment for dealing with the difficult airway should be readily available prior to anesthetic induction [18]. If difficulties are predicted based on the airway examination, general anesthesia can be induced by the inhalation of sevoflurane in 100% oxygen with the maintenance of spontaneous ventilation or as was done in our case, effective bag-valve-mask ventilation demonstrated prior to the administration of a neuromuscular blocking agent. Infiltration of lower airway structures has been reported including tracheal narrowing and vocal cord involvement suggesting that attention should be direct toward ensuring that an appropriate leak is present prior to inflation of the cuff of the endotracheal tube [19]. Given the surgical procedure involved, we choose to use an LMA to manage the airway and avoid the need for endotracheal intubation.
Various respiratory manifestations have been noted in patients with ML II including prolonged or recurrent respiratory infections, which are common due to infiltration of the proximal and distal airway. In addition to the issues related to the airway, the baseline respiratory status of these children may be compromised due to skeletal muscle hypotonia, a narrowed thoracic cage, and recurrent chest infections. This involvement results in primarily a restrictive pattern on pulmonary function testing [20]. Prolonged surgical procedures may require postoperative respiratory support. These patients may also require chronic management of respiratory compromise with techniques such as continuous positive pressure or tracheotomy with ventilator support from an early age [4]. Given the potential effects of the residual effects of anesthetic agents on upper airway and respiratory function, short acting anesthetic agents may be optimal and effective reversal of neuromuscular blockade is mandatory prior to tracheal extubation. In our patient, a brief period of neuromuscular blockade was required for closed hip reduction. To ensure adequate reversal and full return of baseline muscular strength, sugammadex was used to reverse the effects of rocuronium [21].
Given the potential for associated heart disease, as was noted in our patient, preoperative echocardiography is suggested to evaluate the anatomy as well as valvular and myocardial function. As these patients age, progressive myocardial involvement may lead to valvular insufficiency, aortic room dilatation and impairment of myocardial function [22]. A single case report also reports the association of an abnormal coronary artery in a patient with ML II [23].
Another consideration for ML II patients is the presence of gastroesophageal reflux disease (GERD). In symptomatic patients, the preoperative administration of metoclopramide and an H2-antagonist should be considered. The risk-benefit ratio of aspiration versus difficulties with airway management must be considered when deciding upon the optimal technique for airway management. At the conclusion of the surgical procedure, tracheal extubation should occur only when airway protective reflexes are present. Our patient did not receive any preprocedure medications for GERD as she was asymptomatic.
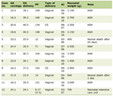
To date, there are only two previous reports of anesthetic care in patients with ML II (Table 1 [14, 15]). When considering the perioperative challenges of such patients, many issues parallel those of patients with MPS including airway, respiratory and cardiac involvement. Skeletal and airway involvement may lead to problems with bag-valve-mask ventilation, LMA placement, and endotracheal intubation using both direct and indirect laryngoscopy. Involvement of the lower respiratory tract predisposes these patients to restrictive lung disease, recurrent respiratory infections, and the potential for perioperative respiratory insufficiency or failure. Prolonged procedures may require postoperative ventilator support. Preoperative echocardiography is suggested to evaluate myocardial and valvular function.
Leroy JG, Demars RI. Mutant enzymatic and cytological phenotypes in cultured human fibroblasts. Science. 1967;157(3790):804-806. doipubmed
Leroy JG, Spranger JW. I-cell disease. N Engl J Med. 1970;283(11):598-599. doi
Leroy JG, Spranger JW, Feingold M, Opitz JM, Crocker AC. I-cell disease: a clinical picture. J Pediatr. 1971;79(3):360-365. doi
Sheikh S, Madiraju K, Qazi Q, Rao M. Improved morbidity with the use of nasal continuous positive airway pressure in I-cell disease. Pediatr Pulmonol. 1998;25(2):128-129. doi
Ishak M, Zambrano EV, Bazzy-Asaad A, Esquibies AE. Unusual pulmonary findings in mucolipidosis II. Pediatr Pulmonol. 2012;47(7):719-721. doipubmed
Sato Y, Kobayashi H, Sato S, Shimada Y, Fukuda T, Eto Y, Ohashi T, et al. Systemic accumulation of undigested lysosomal metabolites in an autopsy case of mucolipidosis type II; autophagic dysfunction in cardiomyocyte. Mol Genet Metab. 2014;112(3):224-228. doipubmed
Carey WF, Jaunzems A, Richardson M, Fong BA, Chin SJ, Nelson PV. Prenatal diagnosis of mucolipidosis II--electron microscopy and biochemical evaluation. Prenat Diagn. 1999;19(3):252-256. doi
Hanai J, Leroy J, O'Brien JS. Ultrastructure of cultured fibroblasts in I-cell disease. Am J Dis Child. 1971;122(1):34-38. doi
Sprigz RA, Doughty RA, Spackman TJ, Murane MJ, Coates PM, Koldovsky O, Zackai EH. Neonatal presentation of I-cell disease. J Pediatr. 1978;93(6):954-958. doi
White KK. Orthopaedic aspects of mucopolysaccharidoses. Rheumatology (Oxford). 2011;50(Suppl 5):v26-33. doipubmed
Peck SH, Casal ML, Malhotra NR, Ficicioglu C, Smith LJ. Pathogenesis and treatment of spine disease in the mucopolysaccharidoses. Mol Genet Metab. 2016;118(4):232-243. doipubmed
Dumontel C, Constant H, Souillet G, Zabot MT. In vitro toxic effects of certain antibiotics on the fibroblasts of two children with I-cell disease. Cell Biol Toxicol. 1998;14(5):333-343. doipubmed
Kempthorne PM, Brown TC. Anaesthesia and the mucopolysaccharidoses: a survey of techniques and problems. Anaesth Intensive Care. 1983;11(3):203-207. pubmed
Mahfouz AK, George G. Anesthesia for gingivectomy and dental extractions in a child with I-cell disease--a case report. Middle East J Anaesthesiol. 2011;21(1):121-124. pubmed
Bains DB, Street N, Overton JH. Anaesthetic implications of mucolipidosis. Paediatr Anaesth. 1993;3(5):303-306. doi
Peters ME, Arya S, Langer LO, Gilbert EF, Carlson R, Adkins W. Narrow trachea in mucopolysaccharidoses. Pediatr Radiol. 1985;15(4):225-228. doipubmed
Wong E, Ng YY. The difficult airway in the emergency department. Int J Emerg Med. 2008;1(2):107-111. doipubmed
Engelhardt T, Weiss M. A child with a difficult airway: what do I do next? Curr Opin Anaesthesiol. 2012;25(3):326-332. doipubmed
Tobias JD. Pediatric airway anatomy may not be what we thought: implications for clinical practice and the use of cuffed endotracheal tubes. Paediatr Anaesth. 2015;25(1):9-19. doipubmed
Lin SP, Shih SC, Chuang CK, Lee KS, Chen MR, Niu DM, Chiu PC, et al. Characterization of pulmonary function impairments in patients with mucopolysaccharidoses--changes with age and treatment. Pediatr Pulmonol. 2014;49(3):277-284. doipubmed
Tobias JD. Current evidence for the use of sugammadex in children. Paediatr Anaesth. 2017;27(2):118-125. doipubmed
Cripe LH, Ware SM, Hinton RB. Replacement of the aortic valve in a patient with mucolipidosis III. Cardiol Young. 2009;19(6):641-643. doipubmed
Siles A, Mitchell GA, Dahdah NS. An infant with mucolipidosis-II and an atretic orifice of the left coronary artery. Cardiol Young. 2010;20(1):97-99. doipubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.