








| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 8, Number 6, June 2017, pages 191-195
A Rare Case of Carney-Stratakis Syndrome in a Patient With SDHA Mutation
Aurelio Negroa, f, Davide Nicolib, Alberto Cavazzac, Rosaria Santia, Stefano Bonilaurid, Enrico Farnettib, Michele Panebiancoe
aInternal Medicine and Hypertension Unit, IRCCS-Arcispedale Santa Maria Nuova Reggio Emilia, Italy
bMolecular Biology Laboratory, IRCCS-Arcispedale Santa Maria Nuova Reggio Emilia, Italy
cPathology Unit, IRCCS-Arcispedale Santa Maria Nuova Reggio Emilia, Italy
dGeneral and Emergency Surgery, IRCCS-Arcispedale Santa Maria Nuova Reggio Emilia, Italy
eOncology Unit, Department of Oncology and Advanced Techmologies, IRCCS-Arcispedale Santa Maria Nuova Reggio Emilia, Italy
fCorresponding Author: Aurelio Negro, Internal Medicine and Hypertension Unit, IRCCS-Arcispedale Santa Maria Nuova Reggio Emilia, Italy
Manuscript accepted for publication May 12, 2017
Short title: Carney-Stratakis Syndrome
doi: https://doi.org/10.14740/jmc2831w
| Abstract | ▴Top |
Paragangliomas (PGLs) and pheochromocytomas (PCCs) are rare tumors that originate from the neuroendocrine tissue along the paravertebral axis. Up to 35% of these tumors may be hereditary either alone or as a component of a multiple tumor syndrome. The gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract. Most GISTs are driven by gain-of-function mutations in KIT (75-80%) or platelet-derived growth factor receptor-α (PDGFRA) (10%). Approximately 15% of GISTs occurring in adults with no KIT or PDGFRA mutations are thermed “wild-type” GISTs. Succinate dehydrogenase (SDH) mutations may contribute to occurrence of wild-type tumors. In 2002, Carney and Stratakis described a new familiar syndrome (CSS) of PGLs/PCCs and GIST caused by germline mutations in SDH. SDH may act as a tumor suppressor gene, and its defects may be oncogenic. In most of CSS cases, the mutations involve the SDHB/C/D subunits; SDHA mutations are reported but extremely rare. We report a case of CSS with a solitary GIST and an incidentally discovered silent PCC harboring an SDHA mutation.
Keywords: Carney-Stratakis syndrome
| Introduction | ▴Top |
Paragangliomas (PGLs) and pheochromocytomas (PCCs) are rare tumors that originate from the neuroendocrine tissue along the paravertebral axis. Up to 35-40% of these tumors may be hereditary either alone or as a component of a multiple tumor syndrome [1]. Over the last decade, there has been progress in understanding the genetic mechanisms involved in the transmission and development of PGLs/PCCs syndromes [2].
The gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract. They are most common in the stomach (50-60%) and small intestine (30-35%) and are less frequent in the colon (5%) and esophagus (< 1%) [3]. Most GISTs are driven by gain-of-function mutations in KIT (75-80%) or platelet-derived growth factor receptor-α (PDGFRA) (10%). However, approximately 15% of GISTs occur in adults with no KIT or PDGFRA mutations; these tumors are termed “wild-type” GISTs. It has been reported that succinate dehydrogenase (SDH) mutations may contribute to occurrence and growth of wild-type tumors [4].
In 2002, Carney and Stratakis described a new familiar syndrome of PGL and gastric stromal sarcoma (CSS) caused by germline mutations in SDH leading to dysfunction of complex II of the electron transport chain. SDH may act as a tumor suppressor gene, and its defects with a reduction in enzymatic activity may be oncogenic. CSS transmission has an autosomal-dominant inheritance pattern, with incomplete penetrance [5]. In most cases, the mutations involve the SDHB/C/D subunits; moreover, germline SDHA mutations are reported but extremely rare [6].
We present a case of CSS with a solitary GIST and an incidentally discovered PCC harboring an SDHA mutation.
| Case Report | ▴Top |
A 56-year-old Caucasian man came to our attention with history of increasing fatigue, vague abdominal discomfort, reflux symptoms and progressive anemia (Hb 8.7 g/dL) starting from 15.3 g/dL one year before. Fecal occult blood result was positive. No past medical history of interest was reported. Physical examination showed a heart rate of 68 beats per minute, blood pressure of 122/80 mm Hg without postural drop and the body mass index was 24.7 kg/m2. The upper gastrointestinal endoscopy revealed a 35-mm gastric sub-mucosal hemorrhaging lesion, suggestive of GIST (Fig. 1). A gastric biopsy was performed and microscopic evaluation showed a well-circumscribed tumor localized in the gastric wall and sparing the mucosa (left upper part of the image). At immunohistochemistry, neoplastic cells were diffusely positive with DOG-1 (c) and CD117/Kit as cells were positive for DOG1, CD117 and negative for desmine and S100 protein. The number of mitoses was small (2 × 50 HPF). Histopathological diagnosis was GIST (Fig. 2a-d).
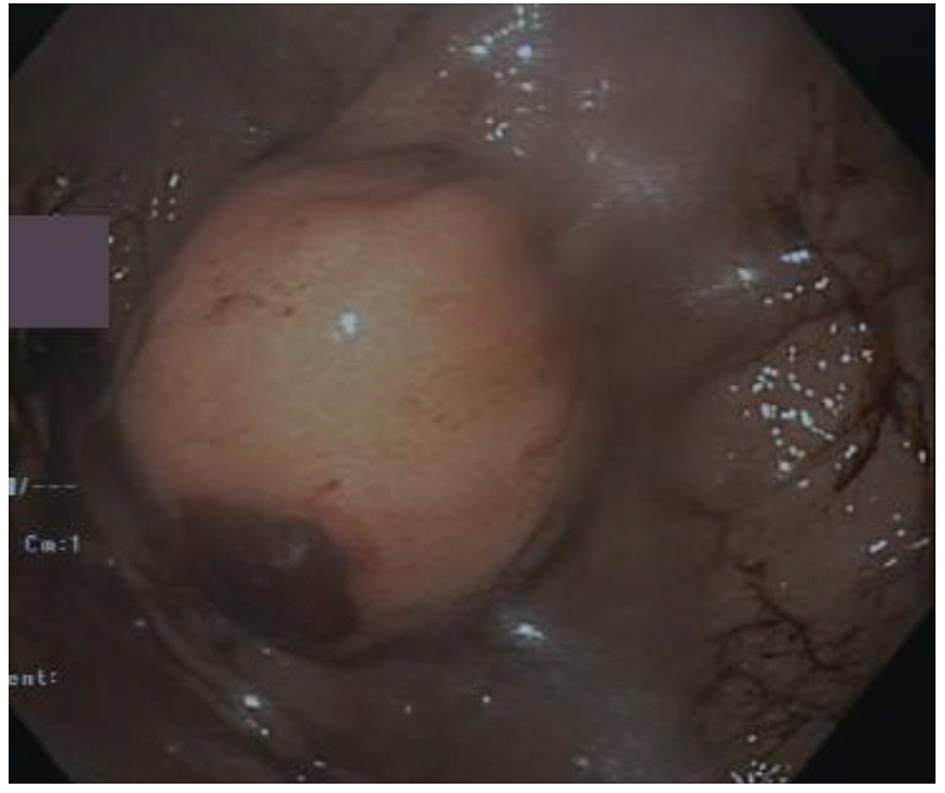 Click for large image | Figure 1. Endoscopy showed a 35-mm gastric sub-mucosal hemorrhaging lesion. |
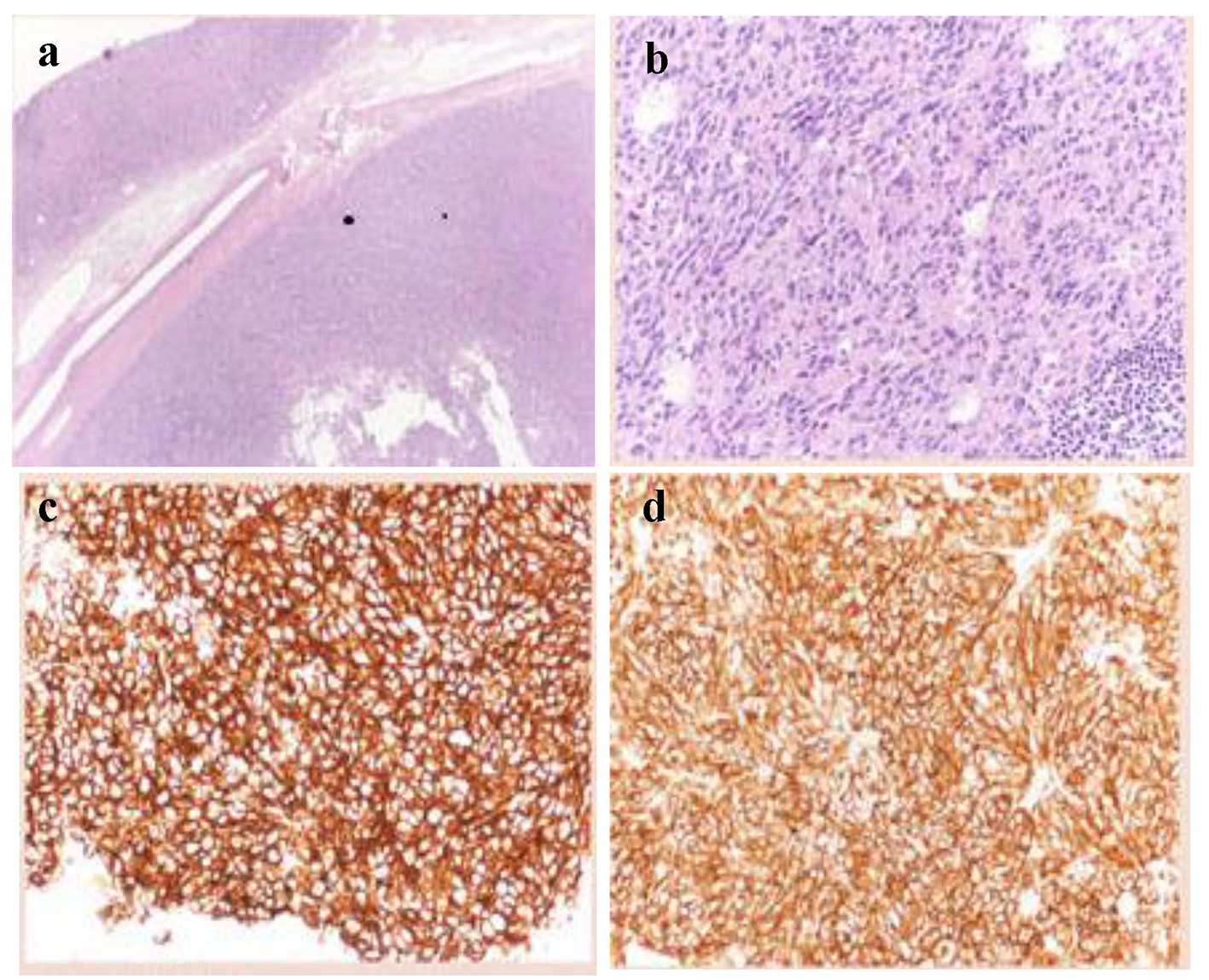 Click for large image | Figure 2. Histologic features of the gastric GIST. (a) Low magnification showing a well-circumscribed tumor localized in the gastric wall and sparing the mucosa (left upper part of the image) (hematoxylin-eosin, × 20). (b) Higher magnification showing a relatively bland proliferation of oval cells with a moderate amount of cytoplasm. Mitoses were inconspicuous. (c, d) At immunohistochemistry, neoplastic cells were diffusely positive with DOG-1 (c) and CD117/Kit. |
A contrast computer tomography (CT) scan of abdomen confirmed the gastric neoplasm and revealed a 10-mm intense (150 HU) and irregular nodule on the right adrenal gland (Fig. 3a). At this point, the patient described infrequent and episodic fast palpitations but denied sweating, headaches or constipation. Moreover, his wife referred mood swings and easy irritability. Patient also referred that his daughter underwent surgery for laterocervical PGL at age of 23 years. Therefore, the adrenal function was investigated. Laboratory findings are shown in Table 1. Notably, repeated 24-h urinary metanephrines measurements were in the normal range. A 18F-flurodeoxyglucose positron emission tomography (PET) showed an intense uptake of the radiotracer at the level of gastric neoplasm and right adrenal gland (SUVmax 33.2 and 8.6, respectively) (Fig. 3b).
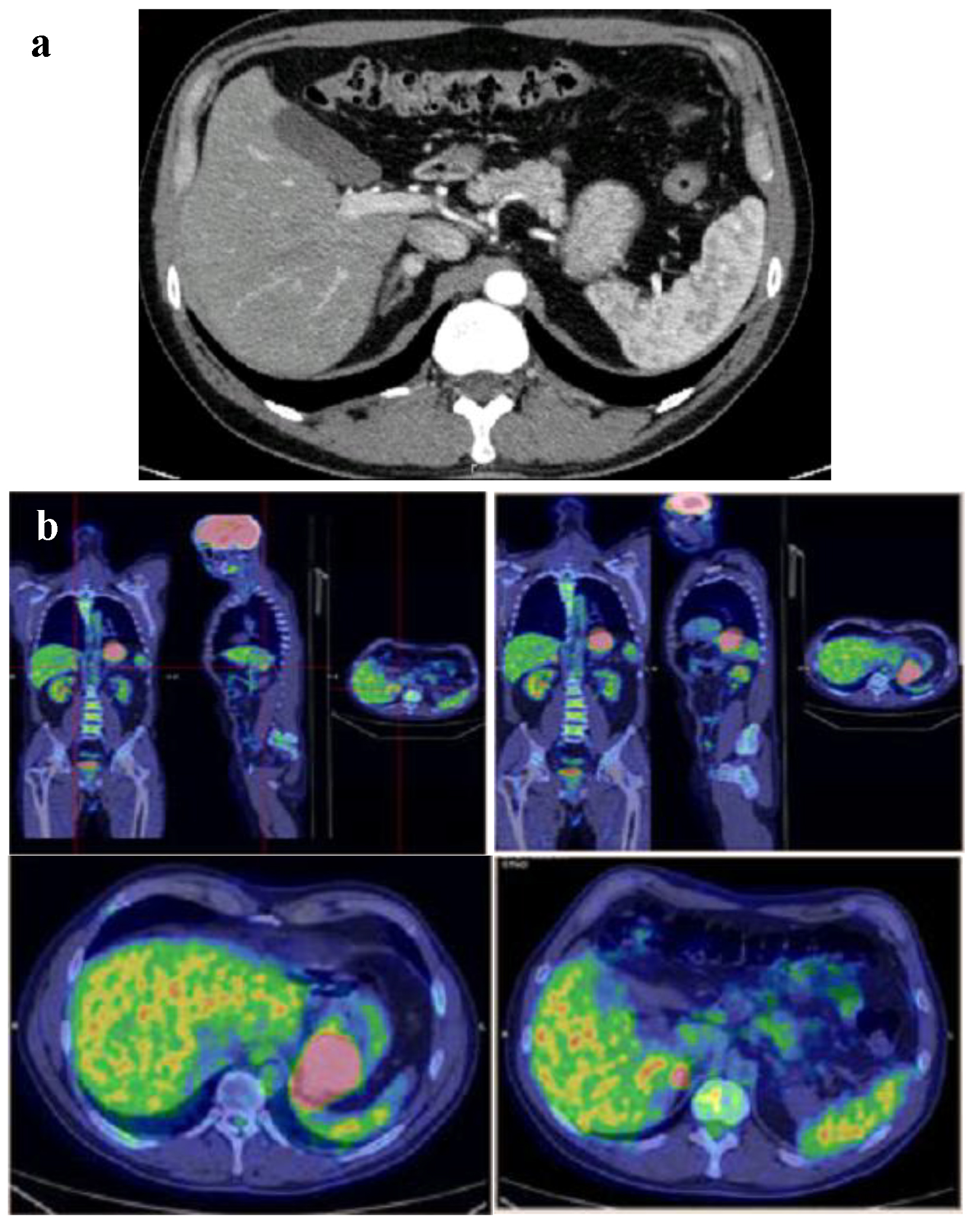 Click for large image | Figure 3. (a) Gastric neoplasm and a 10-mm intense (150 HU) nodule on the right adrenal gland at contrast CT scan of abdomen. (b) Intense uptake of the radiotracer at the level of gastric neoplasm and right adrenal gland at 18FDG-PET. |
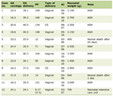 Click to view | Table 1. Preoperative Laboratory Findings |
Patient was treated with doxazosin (4 mg twice daily) and atenolol (25 mg once daily) and underwent laparotomy with radical GIST resection and right adrenalectomy. Histopathology confirmed the diagnosis of GIST, WHO prognostic group 2; histological diagnosis of the adrenal nodule was of PCC, PASS score 4 (Fig. 4). The successive postoperative course was uneventful and the patient was discharged in good clinical condition without specific treatment. The presence of GIST and PCC led us to the suspect of CSS and then genetic analysis was performed. We analyzed the DNA of the patient with NGS approach using Trusight One Sequencing Panel, developed by Illumina, focused on the exonic regions harboring disease causing mutations. We identified a missense mutation of SDHA with A/T variant and a single amino acid substitution (Y/F) at position 629. This mutation was confirmed by allele specific PCR and Sanger sequencing (Fig. 5).
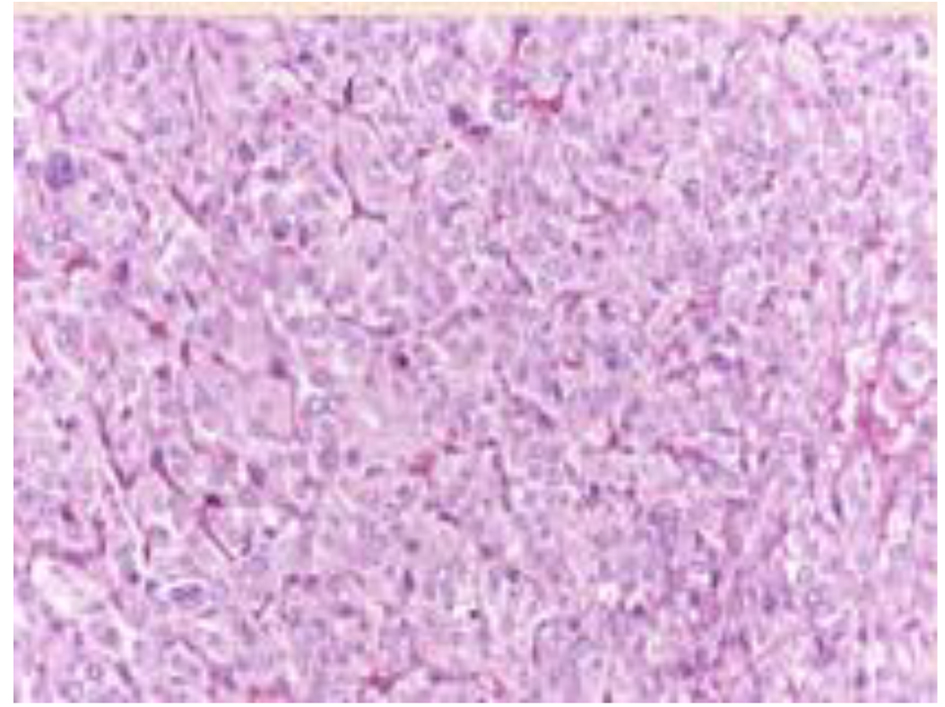 Click for large image | Figure 4. Histologic features of the adrenal pheochromocytoma, consisting in mildly atypical epithelioid cells disposed in well vascularized nests. |
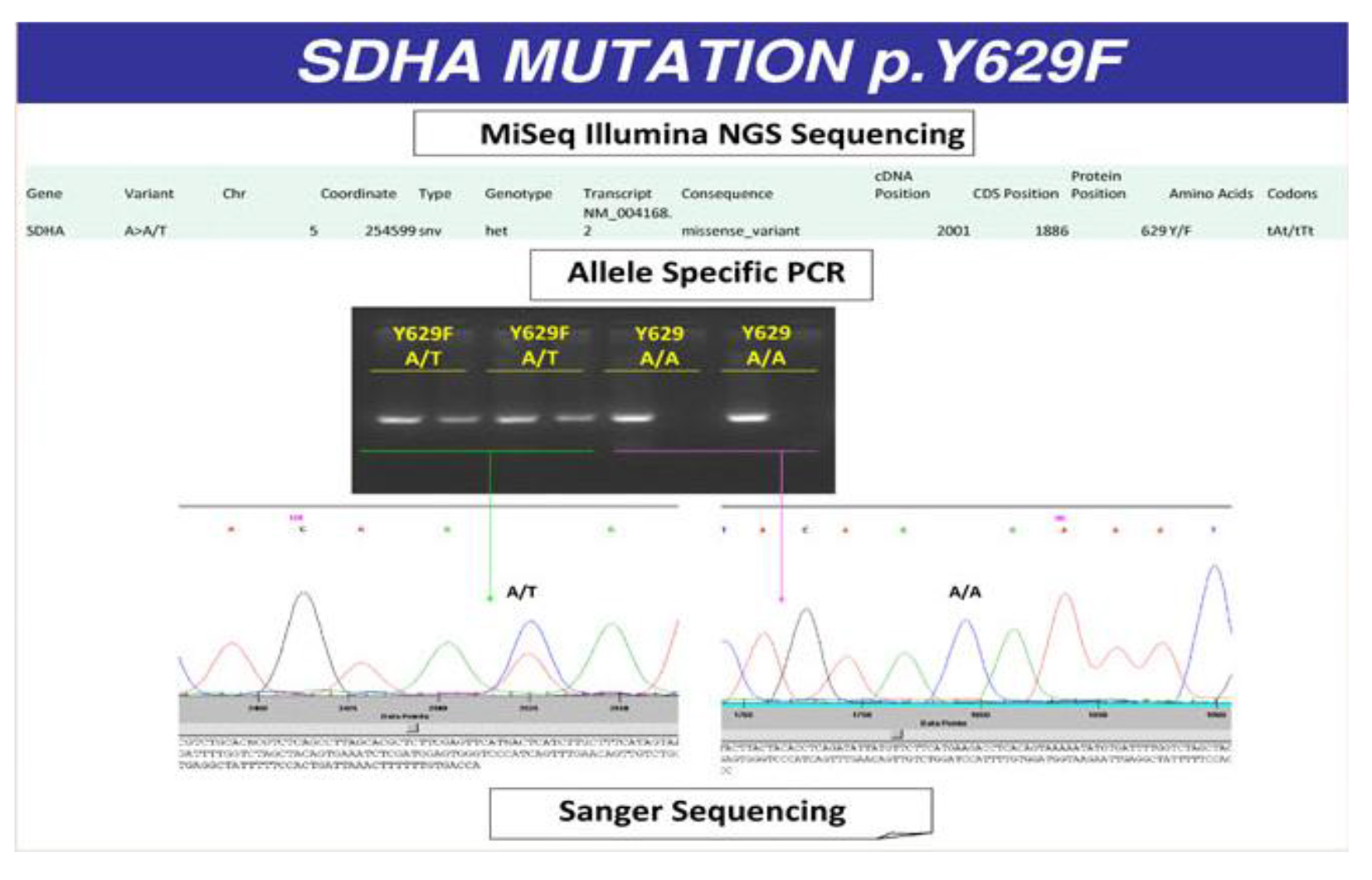 Click for large image | Figure 5. Missense mutation of SDHA with A/T variant and a single amino acid substitution (tyrosine/phenylalanine) at position 629. |
At the follow-up after 12 months, the patient was in a healthy state. We repeated the hormonal tests which were still all in the normal range. No recurrence was detected by CT scan.
The patient signed an informed consent to participate in this case report.
| Discussion | ▴Top |
This patient represents a new case of CSS with a putative functionally relevant mutation in SDHA gene.
PCCs and PGLs are rare neural crest tumors derived from catecholamine-secreting cells of the adrenal medulla or extra-adrenal sympathetic paraganglia, respectively. PCCs produce, store, metabolize and secrete catecholamines or their metabolites; however, extra-adrenal PGLs are usually not associated with catecholamines secretion. The main signs and symptoms of excess circulating catecholamines are headache, palpitations, sweating, sustained or paroxysmal hypertension, orthostatic hypotension, pallor or flushing, nausea, constipation, fatigue, weight loss, anxiety, fever, and hyperglycemia. According to the degree of catecholamines excess, patients may present with hypertensive crisis, myocardial infarction, arrhythmia, acute left ventricular failure, stroke, aortic dissection, and sudden death. However, in the last years, the patients are often diagnosed with an incidentally “asymptomatic” adrenal mass detected by abdominal imaging performed for other reasons [7]. In our experience, such as in this case, when a focused history is obtained, some of the classic symptoms of PCCs/PGLs are often disclosed retrospectively. Non-functioning PCCs, as our case, are uncommon [8, 9] and their coexistence with other neuroendocrine tumors is rarely described in the medical literature. The majority of PCCc/PGLs occur sporadically but, nowadays, up to 35% of cases are attributable to hereditary germline mutations in several genes among which the SDHx complex [1, 2, 7]. Familial PCCs/PGLs are usually transmitted as an autosomal dominant trait, so the offspring of a mutation carrier will have a 50% chance to having inherited the relevant gene mutation. Therefore, many authorities recommend genetic testing in all cases of PCCs/PGLs [10].
GISTS arise from stem cells with characteristics of interstitial cells of Cajal, the pacemaker cells in the digestive tract that regulate the peristalsis. Most GISTs are driven by gain-of-function mutations in KIT (75-80%) or PDGFRA (10%). However, about 15% of GISTs occurring in adults lack KIT or PDGFRA mutations, and they are termed “wild-type” GISTs. It has been reported that SDH mutations may contribute to these so-called “wild-type” tumors [4]. SDH is a four-subunit (A, B, C, and D) Krebs cycle enzymatic complex, located in the inner mitochondrial membrane but encoded by chromosomal DNA. Whatever subunit is changed, the entire complex is hampered, impairing succinate-to-fumarate conversion. Succinate accumulation decreases degradation of hypoxia-inducible factor-1α (HIF-1α); HIF-1α can thus translocate into the nucleus initiating tumorigenic transcription [11].
The occurrence of two or more distinct types of NETs in an individual patient is unusual, except in patients with hereditary syndromes such as von Hippel-Lindau disease, neurofibromatosis 1, mutations in SDH, and multiple endocrine neoplasia. In 2002, Carney and Stratakis described a new familial syndrome of PGL and gastric stromal sarcoma (CSS) caused by germline mutations in SDH with an autosomal-dominant inheritance pattern and incomplete penetrance [5]. In most cases, the mutations involve the SDHB/C/D subunits; SDHA mutations are significantly unreported [6]. Our case is relevant because it presented as isolated hemorrhaging gastric GIST and an incidentally discovered adrenal mass, suspected for PCC at imaging, but without clinical symptoms and laboratoristic signs of adrenergic hyperactivity. The subsequent acknowledgment of a PGL in a daughter corroborated the suspicious of the syndromic nature of GIST. Our genetic analysis demonstrated a missense mutation of SDHA gene with A/T modification in heterozygosity that determined a single amino acid substitution (Y/F) at position 629 in exon 14.
We acknowledge that confirmation of the pathogenicity of this mutation would necessitate transcript analysis and functional studies even so unfeasible in our laboratory. However, we are confident that the SDHA mutation may be relevant considering the two rare neoplasms coexistence in our patient and an analogue tumor in a young daughter.
The natural history of PGLs/PCCs-GIST syndrome is not completely established and there are no data regarding the long-term prognosis of this condition. According to Carney and Stratakis [5], the clinical approach to management of the tumors should be based on the known behavior in other syndromes in which they appear.
In summary, in this report, we have reported another rare case of CSS, with a gastric GIST and a non-functioning and incidentally discovered adrenal PCC, relevant for a new SDHA mutation. We believe that for all patients who present GIST and PCC/PGL, the diagnosis of CSS should be considered to make a genetic evaluation.
| References | ▴Top |
- Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14(2):108-119.
doi pubmed - Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101-111.
doi pubmed - Joensuu H, Vehtari A, Riihimaki J, Nishida T, Steigen SE, Brabec P, Plank L, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
doi - Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108(1):314-318.
doi pubmed - Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108(2):132-139.
doi pubmed - Ricci R. Syndromic gastrointestinal stromal tumors. Hered Cancer Clin Pract. 2016;14:15.
doi pubmed - Pappachan JM, Raskauskiene D, Sriraman R, Edavalath M, Hanna FW. Diagnosis and management of pheochromocytoma: a practical guide to clinicians. Curr Hypertens Rep. 2014;16(7):442.
doi pubmed - Plouin PF, Gimenez-Roqueplo AP. Pheochromocytomas and secreting paragangliomas. Orphanet J Rare Dis. 2006;1:49.
doi pubmed - Sundahl N, Van Slycke S, Brusselaers N. A rare case of clinically and biochemically silent giant right pheochromocytoma: case report and review of literature. Acta Chir Belg. 2016;116(4):239-242.
doi pubmed - Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942.
doi pubmed - Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648-657.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.