








| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 7, Number 8, August 2016, pages 326-330
A Case of Kartagener Syndrome With Pulmonary Hypertension
Babaji Ghewadea, c, Smaran Cladiusa, Swapnil Chaudharia, Arvind Bhakea
aDepartment of Respiratory Medicine, Jawaharlal Nehru Medical College, Sawangi Meghe, Wardha, Maharashtra, India
bDepartment of Pathology, Jawaharlal Nehru Medical College, Sawangi Meghe, Wardha, Maharashtra, India
cCorresponding Author: Babaji Ghewade, Department of Respiratory Medicine, Jawaharlal Nehru Medical College, Sawangi Meghe, Wardha, Maharashtra 442001, India
Manuscript accepted for publication January 08, 2016
Short title: Kartagener Syndrome
doi: http://dx.doi.org/10.14740/jmc2412w
| Abstract | ▴Top |
Primary ciliary dyskinesia (PCD) is a genetically heterogeneous syndrome caused by defect in motile cilia. The true prevalence is unknown but estimated to affect 1:20,000 to 1:100,000 people. Estimated incidence is 1 per 10 to 20,000 births. In 1933, Kartagener described the PCD syndrome as the triad of situs inversus, bronchiectasis, and either nasal polyps or recurrent sinusitis, while the description by Afzelius in 1976 of the defects in the ultrastructure of ciliary dynein arms revealed the basis of this condition. Thus, clinical findings include respiratory distress in neonates, recurrent respiratory tract infections, bronchiectasis, situs inversus, infertility, and heterotaxy in approximately 50%. Here we report a rare case of a young female with recurrent respiratory symptoms, dextrocardia, and situs inversus, who was misdiagnosed as a case of pulmonary tuberculosis and had received three courses of anti-tubercular therapy since year 2003, and finally diagnosed as Kartagener syndrome with severe pulmonary hypertension.
Keywords: Primary ciliary dyskinesia; Kartagener’s syndrome; Situs inversus; Bronchiectasis
| Introduction | ▴Top |
Primary ciliary dyskinesia (PCD) is a genetically heterogeneous syndrome caused by defect in motile cilia [1]. The true prevalence is unknown but estimated to affect 1:20,000 to 1:100,000 people [2]. Estimated incidence is 1 per 10 to 20,000 births. In 1933, Kartagener described the PCD syndrome as the triad of situs inversus, bronchiectasis, and either nasal polyps or recurrent sinusitis, while the description by Afzelius in 1976 of the defects in the ultrastructure of ciliary dynein arms revealed the basis of this condition. Thus, clinical findings include respiratory distress in neonates, recurrent respiratory tract infections, bronchiectasis, situs inversus, infertility, and heterotaxy in approximately 50% [3, 4].
| Case Report | ▴Top |
A 26-year-old lady was admitted with chief complaints of breathlessness of MMRC grade III, right-sided chest pain and cough with expectoration since 1 month. Patient had history of pulmonary tuberculosis for which she had taken irregular courses of anti-tubercular therapy (ATT) thrice in last 10 years.
On examination, patient was underweight with body mass index (BMI) of 18.5 kg/m2. There was grade II clubbing, but no pallor, icterus, cyanosis, lymphadenopathy or pedal edema. Her pulse was 92/min, regular, normovolemic and peripheral pulses were felt bilaterally. Respiratory rate was 22/min, thoracoabdominal, blood pressure was 140/80 mm Hg in right arm in supine position and oxygen saturation was 65% on room air.
Upper respiratory tract examination showed widening of maxillary crest with mucoid secretions discharging from sinuses. In lower respiratory tract, trachea was central, and normal vesicular breath sounds with bilateral crepitations were heard over lower lung fields. Heart sounds were prominently heard on right side and second heart sound was loud.
Chest X-ray posterior-anterior (Fig. 1) view was suggestive of cystic bronchiectasis in both lower zones, more prominent on right side. There was dextrocardia with gastric air bubble on right side and homogeneous opacity below left hemidiaphragm suggestive of situs inversus. Her lab investigations showed hemoglobin 15.5g/dL, WBC 5,400/mm3 with 86% neutrophils and normal platelet counts. Liver and kidney function tests were normal along with normal blood sugar and ELISA test for HIV was non-reactive.
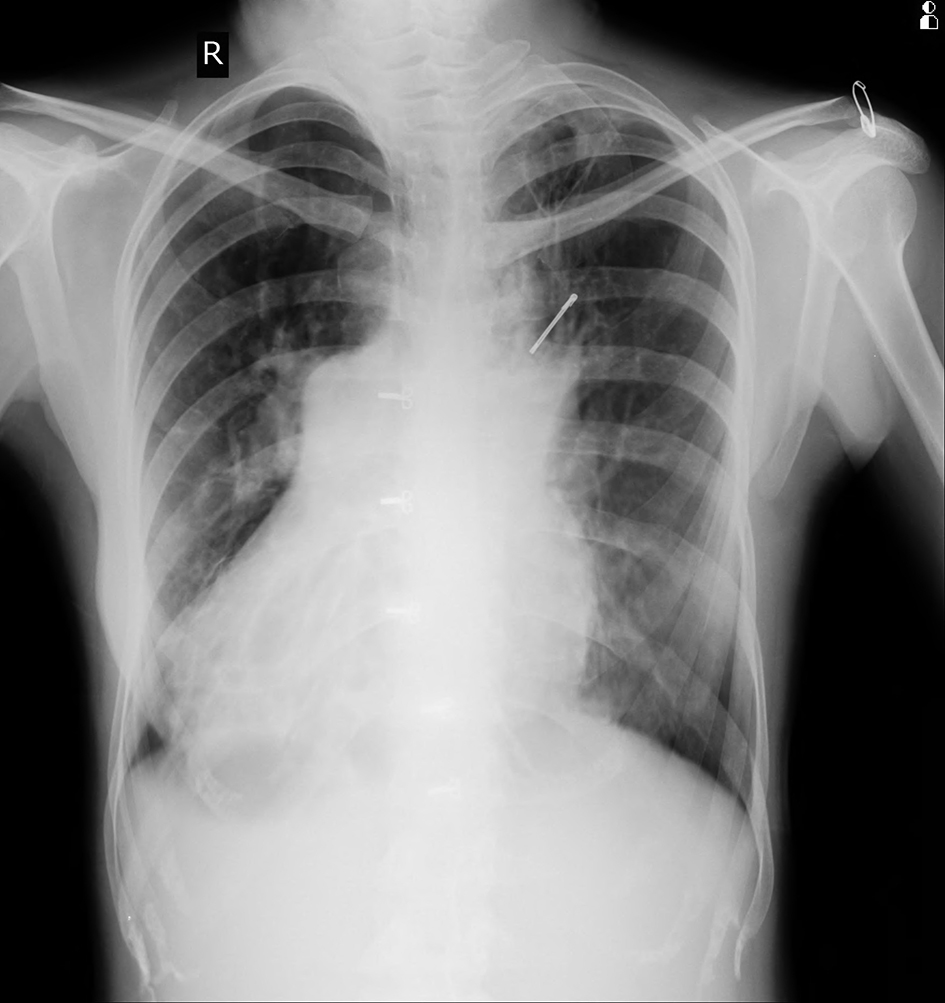 Click for large image | Figure 1. Chest X-ray showing B/L bronchiectasis with dextrocardia. |
HRCT thorax (Fig. 2) showed complete reversal of heart and great vessels suggestive of dextrocardia. The visualized abdominal organs in CT thorax showed stomach and spleen on right side and liver on left side suggestive of complete situs inverses. There was cystic bronchiectasis with ground glass opacities and interlobular septal thickening was noted in left apical and right lower lobe of lungs. Traction bronchiectasis was noted in left lower lobe. Left lung showed decreased volume.
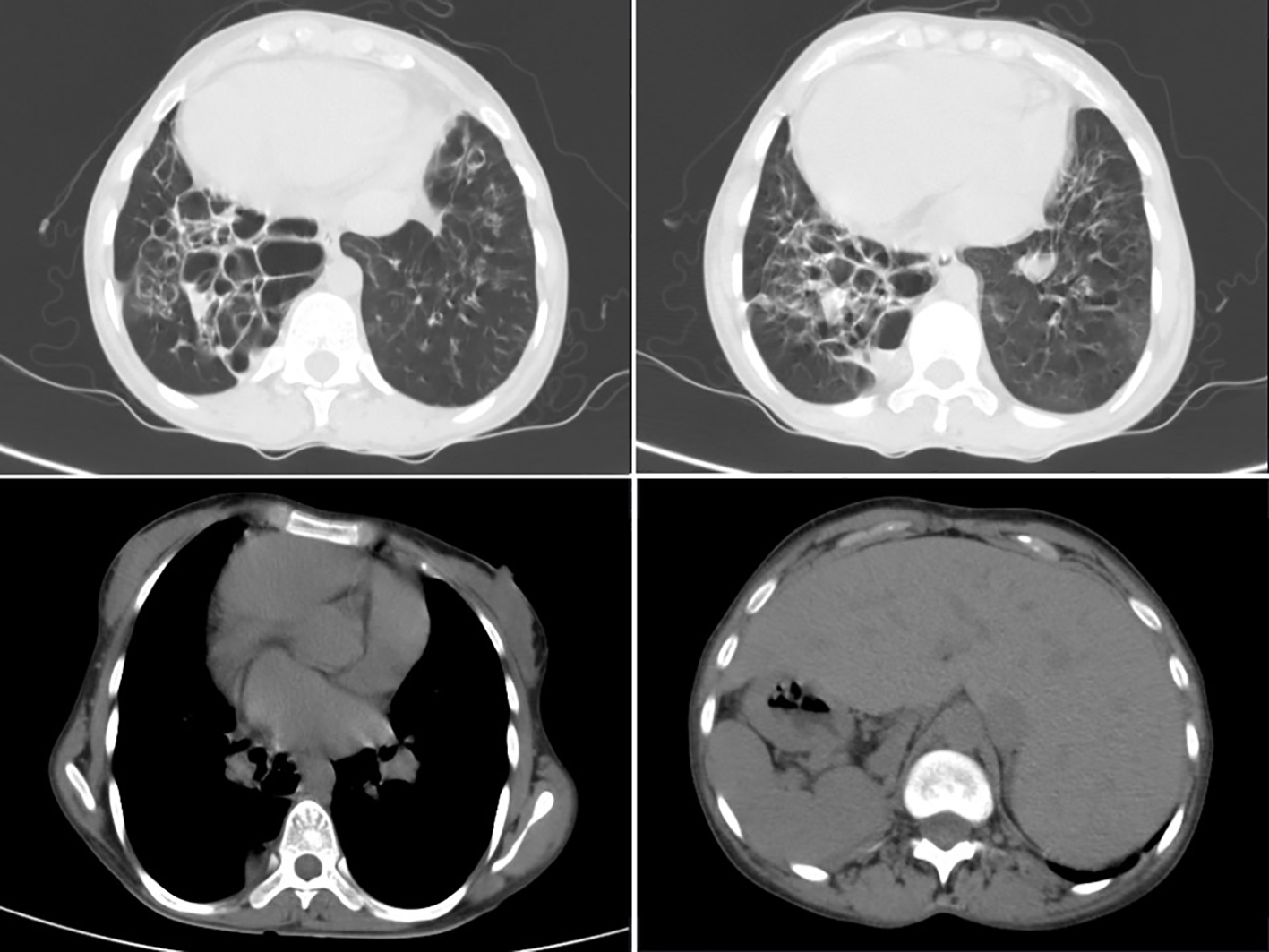 Click for large image | Figure 2. HRCT thorax showing B/L cystic bronchiectasis with dextrocardia with ground glass opacities with evidence of situs inversus. |
USG abdomen and pelvis (Fig. 3a, b) also confirmed complete situs inversus. X-ray waters view and CT PNS (Fig. 4) showed absent frontal sinuses. ECG showed right axis deviation, inverted P wave in lead I and avL, and T wave inversion from v1 to v6 suggestive of dextrocardia. 2D ECHO showed dextrocardia, mild tricuspid regurgitation and severe pulmonary hypertension (PAP 90 mm Hg).
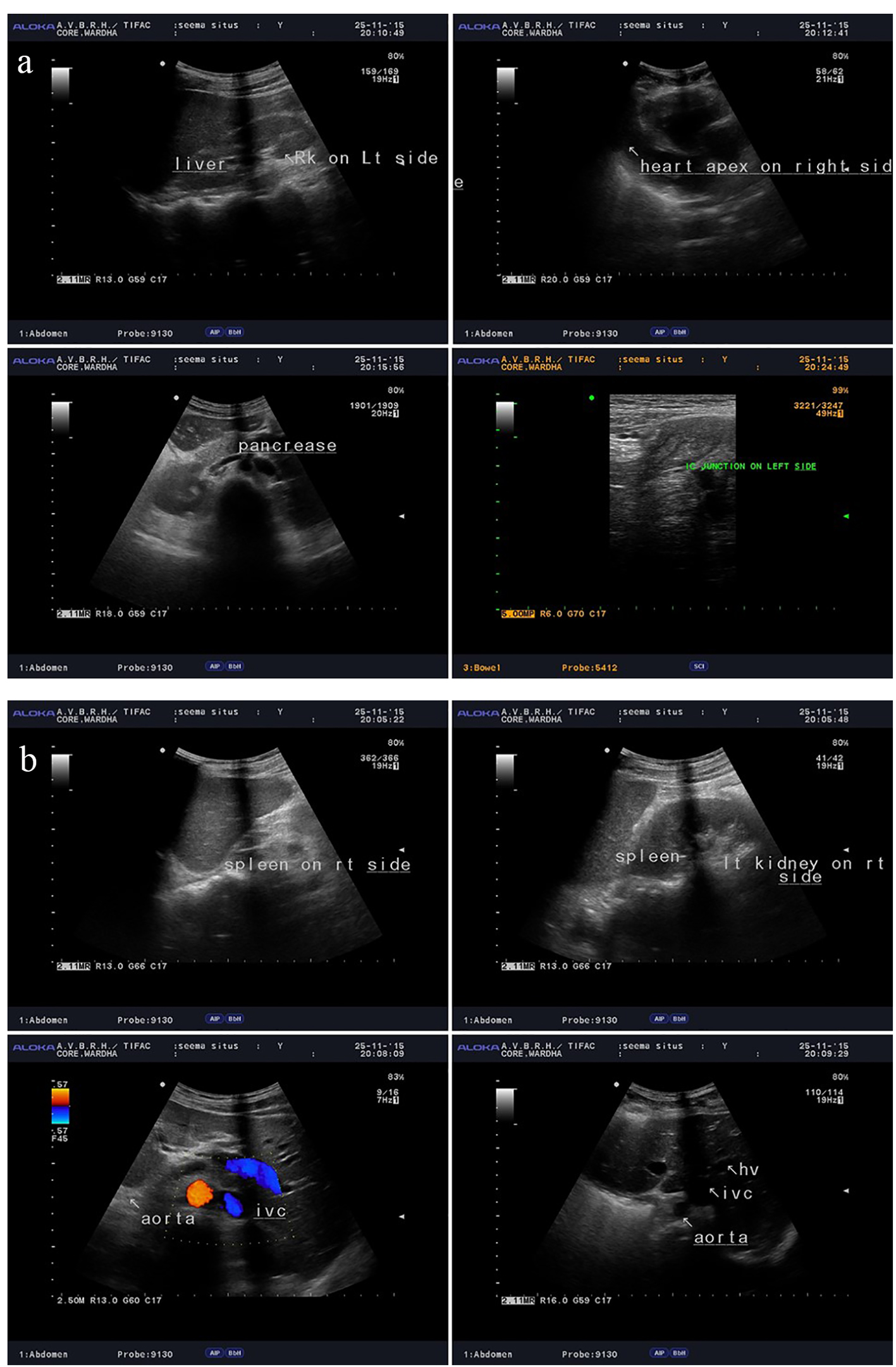 Click for large image | Figure 3. (a, b) USG abdomen and pelvis showing complete situs inversus. |
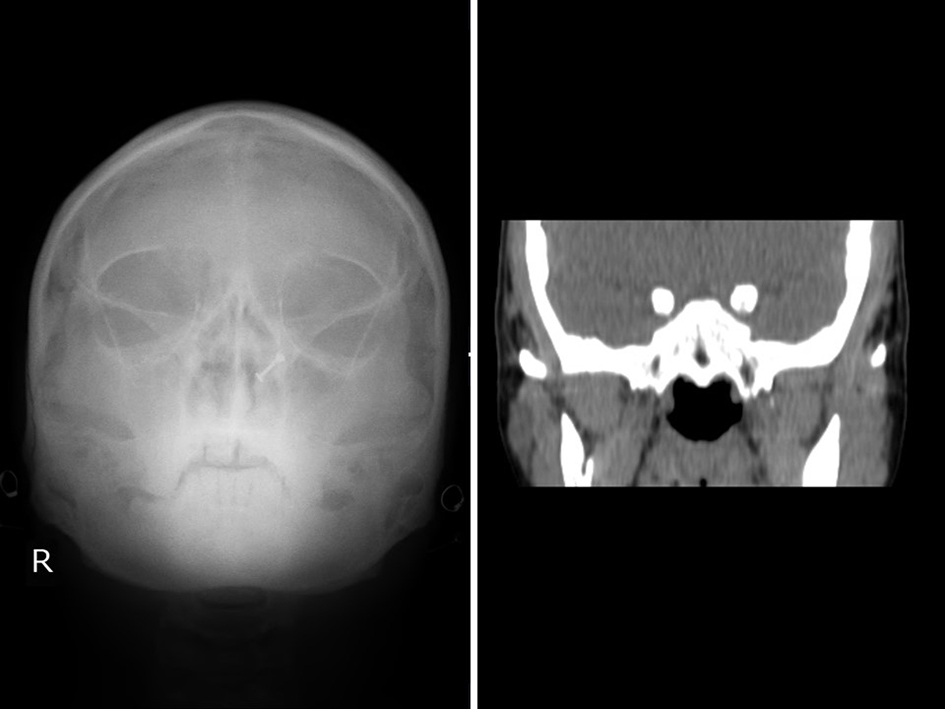 Click for large image | Figure 4. X-ray waters view and CT PNS showing absent frontal sinus. |
Saccharin test showed detection of sweetness at 2 h and 17 min. This confirmed impaired nasal mucociliary clearance. Nasal scrape cytology for demonstration of cilia (Fig. 5) showed multiple groups of columnar cells which were ciliated. Also seen are cell groups which were cuboidal along with few inflammatory cells with degeneration and scanty hemorrhagic material and mucin. No dysplastic pathology was seen.
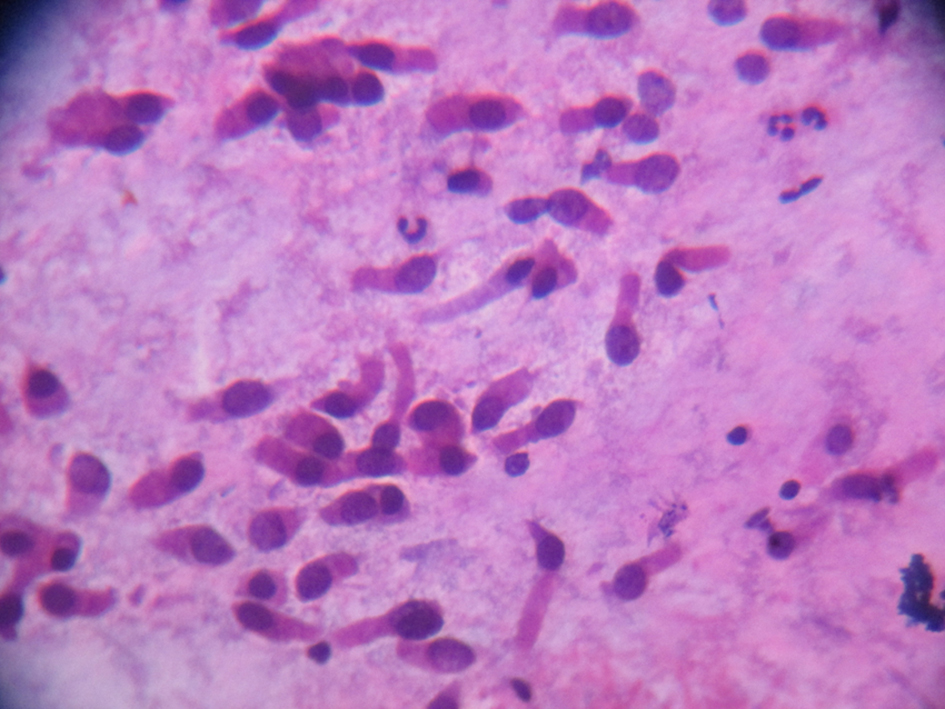 Click for large image | Figure 5. Ciliated columnar epithelium from nasal scrapping. |
Patient was managed with supplemental oxygen, antibiotics, bronchodilators and mucolytic agents.
| Discussion | ▴Top |
Kartagener syndrome was first described in 1904 by Siewert and then in 1935 by Kartagener as the triad of situs inversus, sinusitis and bronchiectasis. Since then, others have also reported cases of Kartagener syndrome [5, 6]. Taniya et al in 1984 observed situs inversus viscerum totalis in a Japanese male cadaver (61-year-old) with chronic sinusitis and bronchiectasis [7]. Tanaka et al in 2007 described similar cases of two sisters (case 1: 25-year-old; case 2: 19-year-old) who had a healthy brother. The saccharin clearance time (SCT) was measured to examine the mucociliary function. In both cases, the SCT lasted over 60 min. Roentgenography and CT scans revealed that both patients had the clinical triad of chronic sinusitis, chronic bronchitis with bronchiectasis, and situs inversus [8]. Recently, Ciancio et al have reviewed eight cases of Kartagener syndrome from 2006 to 2014. However, two patients with more severe clinical behavior died before 2014 [9]. Arunabha et al also reported a classical case of Kartagener syndrome [10].
Kartagener syndrome may be either situs solitus in which case only dextrocardia is present or situs inversus totalis where all the visceral organs are on the opposite side. The patient in the present report had situs inversus totalis [11]. The clinical features are a result of impaired mucociliary clearance from the upper and lower respiratory tracts, the paranasal sinuses and the middle ear. Male infertility is also usually present, although this need not be the case where mucociliary transport is defective because of random ciliary orientation rather than immotility, so that men with PCD should have seminal analysis before being told that they are infertile. Dextrocardia occurs in about half of cases in the absence of other congenital anomalies. Respiratory symptoms date from infancy or early childhood and include chronic rhinorrhea and sinusitis. There may be otitis media and mild deafness. Cough is prominent and is indeed essential to compensate for the absence of normal mucociliary transport by shifting otherwise stagnant mucus. Recurrent lower respiratory tract infection may result in bronchiectasis, which is therefore acquired, because of delay in the removal of infected material from the bronchi [4].
Cilia are hair-like attachments found on the epithelial surfaces (200 per cell) of various organs. Each cilium is composed of approximately 250 proteins organized into longitudinal microtubules, which make up the basic axonemal structure. Based on the arrangement of the microtubules, cilia are classified into motile cilia, primary cilia and nodal cilia. Motile cilia are the cilia found in the apical surfaces of the upper and lower respiratory tract, the ependymal cells lining the ventricles of the central nervous system, the oviducts of the female reproductive tract and the flagellum of the male sperm. Defects in the nodal cilia may cause errors with left-right body orientation, for example, dextrocardia, situs inversus totalis and situs ambiguous. This explains the association of organ laterality defects in PCD [12].
Motile cilia are organized into nine microtubule pair doublets, surrounding a central pair creating a distinctive 9 + 2 arrangement. The central pair is linked to the surrounding pair doublet through an array of radial spoke proteins and the surrounding pair doublets are linked to one another via nexin-linked proteins. Through ATP-containing dynein arms on the peripheral microtubules, the microtubules slide by one another to produce ciliary motion. The protein links between the microtubules limit the degree of sliding and allow the cilium to bend. Dyneins can be subdivided into axonemal and cytoplasmic dyneins. Axonemal dyneins move cilia and flagella, as described previously, while cytoplasmic dyneins are involved in the organization of spindle poles during mitosis. Axonemal dyneins form two structures, the inner and outer arms, and are attached to the microtubules of the nine outer doublets throughout the length of the axoneme, thus they are central to the process of the bending of the cilium or sperm tail [12].
The diagnosis should be considered in any patient with a history of recurrent upper and lower respiratory tract infection since childhood and can be regarded as established if 1) the features of Kartagener’s syndrome are present, 2) an adult male gives a consistent respiratory history and is found to have immotile live sperms or 3) a woman or child gives a consistent respiratory history and has a sibling with Kartagener syndrome or has consistent ultrastructural defects on a nasal or bronchial epithelial brush biopsy. Impaired mucociliary clearance may be detected in adults by the presence of an abnormal saccharin test, in which the time is recorded for saccharin to be tasted in the mouth after a 0.5-mm particle has been deposited on the inferior turbinate in the nose. This usually takes about 30 min but times of up to 1 h may be normal [13].
Pulmonary hypertension in Kartagener syndrome can be due to bronchiectasis (chronic lung disease and/or hypoxia) and can vary from mild to severe form [14].
Conclusion
It is advised to consider the possibilities of Kartagener syndrome in patients presenting with recurrent upper and lower respiratory tract infections, sinusitis or bronchiectasis with dextrocardia. There are no specific guidelines for the treatment of Kartagener syndrome. Barbato et al compiled existing evidence to formulate general clinical recommendations which include at least biannual clinical visits with routine spirometry, sputum culture and if needed imaging studies [15]. It was found that antibiotic treatment was effective for exacerbations. New studies have supported prophylactic therapy, with gentamicin demonstrating decreased exacerbation frequency [16].
Conflicts of Interest
None.
Financial Disclosure
None.
| References | ▴Top |
- Leigh MW, Pittman JE, Carson JL, Ferkol TW, Dell SD, Davis SD, Knowles MR, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. 2009;11(7):473-487.
doi pubmed - Zariwala MA, Knowles MR, Omran H. Genetic defects in ciliary structure and function. Annu Rev Physiol. 2007;69:423-450.
doi pubmed - Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193(4250):317-319.
doi pubmed - Eliasson R, Mossberg B, Camner P, Afzelius BA. The immotile-cilia syndrome. A congenital ciliary abnormality as an etiologic factor in chronic airway infections and male sterility. N Engl J Med. 1977;297(1):1-6.
doi pubmed - Siewert A. U bereinen Fall von BronchiektasiebeieinemPatientenmitSitus inversusviscerum. Berl Klin Wochenschr. 1904;41:139-141.
- Kartagener MI. Mitteilung: BronchiektasienbeiSitus visceruminversus. BeitrKlinTuberk. 1935;83:489-501.
- Taniya S, Ohmachi N, Akita M, Ohsaki R, Kaneko K. [A case of situs inversus viscerum totalis associated with various abnormalities]. Kaibogaku Zasshi. 1984;59(2):94-103.
pubmed - Tanaka K, Sutani A, Uchida Y, Shimizu Y, Shimizu M, Akita M. Ciliary ultrastructure in two sisters with Kartagener's syndrome. Med Mol Morphol. 2007;40(1):34-39.
doi pubmed - Ciancio N, de Santi MM, Campisi R, Amato L, Di Martino G, Di Maria G. Kartagener's syndrome: review of a case series. Multidiscip Respir Med. 2015;10(1):18.
doi pubmed - Arunabha DC, Sumit RT, Sourin B, Sabyasachi C, Subhasis M. Kartagener's syndrome: a classical case. Ethiop J Health Sci. 2014;24(4):363-368.
doi pubmed - Noone PG, Bali D, Carson JL, Sannuti A, Gipson CL, Ostrowski LE, Bromberg PA, et al. Discordant organ laterality in monozygotic twins with primary ciliary dyskinesia. Am J Med Genet. 1999;82(2):155-160.
doi - Lobo LJ, Zariwala MA, Noone PG, Ciliary dyskinesias: primary ciliary dyskinesia in adults. Eur Respir Mon. 2011;52:130-149.
doi - Afzelius BA. Genetics and pulmonary medicine. 6. Immotile cilia syndrome: past, present, and prospects for the future. Thorax. 1998;53(10):894-897.
doi - Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-41.
doi pubmed - Barbato A, Frischer T, Kuehni CE, Snijders D, Azevedo I, Baktai G, Bartoloni L, et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur Respir J. 2009;34(6):1264-1276.
doi pubmed - Bush A, Chodhari R, Collins N, Copeland F, Hall P, Harcourt J, Hariri M, et al. Primary ciliary dyskinesia: current state of the art. Arch Dis Child. 2007;92(12):1136-1140.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.