








| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 2, Number 2, April 2011, pages 67-70
Arginase Deficiency Presenting as Intractable Hiccups With Severe Hyperammonemia at One Month of Age
Shigeo Iijima
Department of Neonatology, Toho University School of Medicine, 6-11-1 Omori-nishi, Ota-ku, Tokyo 143-8541, Japan
Manuscript accepted for publication February 4, 2011
Short title: Arginase Deficiency With Severe Hyperammonemia
doi: https://doi.org/10.4021/jmc141w
| Abstract | ▴Top |
Arginase deficiency is a rare urea cycle disorder and generally presents in children of preschool age with developmental delay and spasticity. Unlikely other urea cycle defects, patients rarely present hyperammonemic symptoms, especially in the early infantile period. A one-month-old male infant was admitted to our hospital because of abnormal respiratory condition with persistent hiccups and lethargy. The initial laboratory studies showed respiratory alkalosis and remarkable hyperammonemia [ammonia, 1970 µmol/L (25 times normal)]. Amino acid chromatography showed a markedly elevated plasma arginine level (1131 µmol/L; 10 times normal), and erythrocyte arginase activity was undetectable. Despite prompt diagnosis and normalization of ammonia and arginine levels by peritoneal dialysis, the patient developed neurodevelopmental impairment during the one-year follow-up period. In summary, this is a rare case of arginase deficiency in terms of early infantile onset and presentation of the severe form of hyperammonemia. Pathologic hiccups could be one of the clinical clues to the presence of hyperammonemia even in young infants with arginase deficiency.
Keywords: Arginase deficiency; Hyperargininemia; Hyperammonemia; Hiccup; Infant
| Introduction | ▴Top |
The urea cycle is comprised of five enzymes and one cofactor producer, and there are six inherited urea cycle disorders (UCDs) [1]. In Japan, the prevalence of UCDs is estimated as 1 per 50,000 live births [2]. Arginase deficiency is the least common of the UCDs. It is inherited autosomal recessively and its prevalence in Japan is estimated as 1 in 2,000,000 living births [2]. The clinical features of this rare disease differ from those of the other UCDs. In general, arginase deficiency does not present in the early infantile period and its symptoms do not usually include severe hyperammonemia [1]. We describe a rare patient with arginase deficiency who presented with persistent hiccups due to severe hyperammonemia at 1 month of age.
| Case Report | ▴Top |
Our patient was a one-month-old boy, the third child of healthy, non-consanguineous parents. His brother died of accidental ingestion at 8 months of age following failure to thrive and developmental delay due to unknown cause. His 5-year-old sister was well. The patient was born at term by normal delivery, with Apgar scores of 9 at 1 and 5 minutes and birth weight of 3544 g. Physical examination at birth was normal. He was exclusively breast-fed. He showed good vitality, maintenance of normal appetite, and good physical growth despite repeated vomiting. At 44 days of age, the infant developed a bad temper and poor suckling from the morning, and in the following evening, he exhibited continuous hiccups and became lethargic. He was brought to our hospital because of abnormal respiratory condition as the chief complaint.
On physical examination, he had no dysmorphic features, and his weight (4815 g), length, and head circumference were appropriate for his age. He had a low-grade fever (37.7 degrees Celsius) and continuous hiccups that synchronized with his breathing (rate 60/min). However, he did not have central cyanosis, abnormal respiratory and heart sounds, nor general convulsive movement. Initial laboratory studies were notable for respiratory alkalosis with compensatory renal adaptation (pH, 7.47; PCO2, 20.1 mmHg; bicarbonate, 14.4 mmol/L; base excess, - 8.6 mmol/L), and a slightly elevated ALT level of 71 U/L. However, his symptom of hiccups and his laboratory data could not be adequately explained by acute infection and therefore a metabolic workup was done. Determination of the plasma ammonia level showed remarkable hyperammonemia of 1970µmol/L (normal range, < 80 µmol/L). An initial diagnosis of hyperammonemia syndrome due to a metabolic disease or Reye’s syndrome was made, and peritoneal dialysis was performed. Over the next 12 hours, the plasma ammonia level nearly normalized to 102 µmol/L. The prolonged hiccups subsided and the patient’s appearance improved. Peritoneal dialysis was continued and diet treatment with a protein-free formula was begun. Urinary organic acid analysis (gas chromatography/mass spectrometry) showed obviously elevated orotate and uracil levels. Amino acid chromatography (high-pressure liquid chromatography) showed markedly elevated plasma arginine (1131 µmol/L; normal range 65 - 98 µmol/L) and mildly elevated plasma glutamate and glutamine levels with increased urinary excretion of glutamate, glutamine, proline, alanine, citrulline, cystine, ornithine, lysine, and arginine (6964 µmol/L; normal range 33 - 99 µmol/L) (Table 1). These findings corroborated a provisional diagnosis of hyperargininemia. Then, diet treatment with arginine-free formula was started, and peritoneal dialysis was terminated in stages by hospital day 15. The plasma ammonia concentration had been controlled under peritoneal dialysis, and after dialysis was discontinued, it fluctuated between 43 and 138 µmol/L. The plasma arginine concentration decreased to 234 µmol/L on hospital day 24 and to 37 µmol/L on the day of discharge, which was 49 days after the initial diagnosis. There was negligible arginase activity in red blood cells (< 1 µmol/h/g hemoglobin; normal range, 4240 ± 1110 µmol/h/g hemoglobin), and arginase deficiency was diagnosed. Computed tomography was performed on the day of discharge and revealed slight cerebral atrophy with enlargement of the sulci and ventricles. Electroencephalogram showed occasional diffuse slow-wave bursts. After discharge, the patient was treated with a protein-restricted diet (natural protein 1 g/kg per day, with arginine daily intake 300 mg/day). However, during the approximately 1-year follow-up period, he developed mild to moderate motor delay; he could hold his head steady at 6 months, roll over at 8 months, and sit without support at 12 months. No spasticity was noted. Examination of arginase activity in red blood cells obtained from his father, mother, and healthy sibling showed reduced arginase activity levels (1802, 1618, and 1360 µmol/h/g hemoglobin, respectively) and delayed patterns in the arginine loading test, suggesting that they were obligatory carriers.
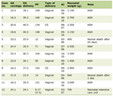 Click to view | Table 1. Pretreatment and 2 Month Posttreatment Amino Acid Levels in Plasma and Urine |
| Discussion | ▴Top |
In most reported cases of arginase deficiency, clinical manifestations are noticed in the preschool or early school years, although retrospective analysis suggested that symptoms may occur in the first year of life, including irritability, inconsolable crying, anorexia, vomiting, self-restricted protein intake, and developmental delay [1]. The clinical picture of our patient differs from those of the majority of previously reported cases by the early infantile onset, marked hyperammonemia, and a curious respiratory symptom of persistent hiccups.
Symptomatic early-onset presentation prior to 3 months of age of arginase deficiency is rare with only 4 reported cases. Picker et al [3] described a female neonate who demonstrated hypertonicity and tachypnea at 30 hours after birth, followed by seizures and cerebral edema, and who died of acute encephalopathy at 3 days of age despite effective hemodialysis. Jorda et al [4] described a male neonate who developed vomiting and hypotonia at 5 days after birth, became comatose with cholestasis, and died at 7 weeks of age. Schiff et al [5]reported a female neonate who manifested respiratory distress followed by drowsiness and seizures at 3 weeks of age, and developed severe encephalopathy despite appropriate but delayed therapy starting from day 47. Braga et al [6] reported a 2-month-old female infant with isolated hepatic presentation and a cholestatic cirrhotic liver who was mentally normal at 2 years of age.
The plasma levels of ammonia, arginine, and glutamine in the above four cases and the present case are shown in Table 2. Compared with the four previously reported cases, the plasma ammonia level in our patient was extremely high. The relationship between hyperammonemia and outcome in UCDs has been reported as follows: the peak blood ammonia level during the first hyperammonemic attack was correlated with neurodevelopmental outcome [7]. When the plasma ammonia concentration exceeded 300 µmol/L initially or 480 µmol/L at its peak, the patient died or had severe neurological deficits [7]. Our case illustrates that hyperargininemia can cause life-threatening hyperammonemia even in the early infantile period and requires early management as usually recommended in other UCDs. On the other hand, the patient described by Picker et al [3] died despite the lower ammonia level. The cerebrospinal fluid (CSF) glutamine level in that patient was very high, and Picker et al described that the elevated CSF (but not blood) glutamine might provide a clue into the pathogenic mechanism leading to brain damage. The blood and urine glutamine levels in our case did not significantly differ from those in the case of Picker et al, but we were unable to assay CSF amino acid levels. The pathogenesis of the neurological disorder in patients with argininemia is not clear. Evidence in the literature is available to support both the hyperammonemia hypothesis and the glutamine hypothesis. To eliminate cerebral ammonia, glutamine enters astrocytes and may alter osmotic regulation followed by brain edema [8, 9]. It remains unclear whether an elevated level of cerebral ammonia or cerebral glutamine is more toxic. The different outcomes might reflect glutamine rather than ammonia, and the relatively low glutamine level in our patient might reflect the relatively improved outcome despite the higher ammonia level.
Click to view | Table 2. Clinical Data in Reported Cases of Arginase Deficiency With Early-Onset (Before 3 Months After Birth) Symptoms |
As for the symptoms at onset, each of the four previously reported patients manifested symptoms unlike those observed in typical cases seen from late infancy. Two of them developed respiratory abnormalities but the details were not described. Our case manifested a curious respiratory condition of intractable hiccups. Hiccups have been characterized as abrupt, inspiratory muscle contractions resulting in brief inspiratory flows terminated by laryngeal closure [10]. There is no clear physiologic purpose of hiccups. Transient hiccupping spells are familiar to all but are usually of little clinical importance. The pathogenesis of hiccups is complex and not fully understood. However, it is known that hiccups in unintubated babies sometimes replace eupneic breathing so that adequate ventilation is achieved by hiccupping only [11], and hyperventilation tends to increase the intensity of hiccups [12]. In our case, hyperventilation may have been self-perpetuating hiccups, and the hiccupping spells may have replaced his breathing. It is said that prolonged hiccupping is generally mediated by a variety of etiologies including some metabolic derangements and leads to respiratory alkalosis in some cases [10, 11]. Respiratory alkalosis is an initial and highly sensitive sign of UCDs with hyperammonemia. Respiratory alkalosis develops due to the fact that ammonia stimulates the respiratory centers of the central nervous system. Therefore, hiccupping spells should be regarded as a nonspecific symptom related to the severity of hyperammonemic encephalopathy.
Despite administering appropriate initial treatment starting on the day of admission with prompt normalization of ammonia and arginine levels in body fluids, our patient developed neurodevelopmental impairment. Arginase deficiency is considered to be a metabolic disease amenable to nutritional therapy. Neonatal screening for arginase deficiency is carried out at some hospitals and has successfully identified affected individuals [13, 14].Detection and treatment with the proper diet soon after birth are warranted to prospectively treat these patients with a view towards amelioration or avoidance of problems.
Conflict of Interest
This report is completely author-owned and has not been funded by any sources. The author has not any conflicts of interest associated with this study. In addition, I did not give any honorarium, grant, or other form of payment to anyone to produce the first draft of the manuscript.
| References | ▴Top |
- Brusilow SW, Horwich AL. Urea cycle enzymes. In: Scriver CR, Beaudet AL, Sly WS, Vogelstein B, Childs B, Kinzler KW (eds.), The metabolic and molecular basis of inherited disease 8th ed., McGraw-Hill, New York, 2001. pp.1909-1963.
- Nagata N, Matsuda I, Oyanagi K. Estimated frequency of urea cycle enzymopathies in Japan. Am J Med Genet 1991;39(2):228-229.
pubmed doi - Picker JD, Puga AC, Levy HL, Marsden D, Shih VE, Degirolami U, Ligon KL, et al. Arginase deficiency with lethal neonatal expression: evidence for the glutamine hypothesis of cerebral edema. J Pediatr 2003;142(3):349-352.
pubmed doi - Jorda A, Rubio V, Portoles M, Vilas J, Garcia-Pino J. A new case of arginase deficiency in a Spanish male. J Inherit Metab Dis 1986;9(4):393-397.
pubmed doi - Schiff M, Benoist JF, Cardoso ML, Elmaleh-Berges M, Forey P, Santiago J, Ogier de Baulny H. Early-onset hyperargininaemia: A severe disorder? J Inherit Metab Dis 2009.
pubmed - Braga AC, Vilarinho L, Ferreira E, Rocha H. Hyperargininemia presenting as persistent neonatal jaundice and hepatic cirrhosis. J Pediatr Gastroenterol Nutr 1997;24(2):218-221.
pubmed doi - Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation.Eur J Pediatr 2003;162(6):410-416.
pubmed - Kanamori K, Ross BD, Chung JC, Kuo EL. Severity of hyperammonemic encephalopathy correlates with brain ammonia level and saturation of glutamine synthetase in vivo. J Neurochem 1996;67(4):1584-1594.
pubmed doi - Albrecht J, Dolinska M. Glutamine as a pathogenic factor in hepatic encephalopathy. J Neurosci Res 2001;65(1):1-5.
pubmed doi - George J, Thomas K, Jeyaseelan L, Peter JV, Cherian AM. Hyponatraemia and hiccups. Natl Med J India 1996;9(3):107-109.
pubmed - Brouillette RT, Thach BT, Abu-Osba YK, Wilson SL. Hiccups in infants: characteristics and effects on ventilation. J Pediatr 1980;96(2):219-225.
pubmed doi - Campbell LA, Schwartz SH. An unusual cause of respiratory alkalosis. Chest 1991;100(4):1159.
pubmed doi - Snyderman SE, Sansaricq C, Norton PM, Goldstein F. Argininemia treated from birth. J Pediatr 1979;95(1):61-63.
pubmed doi - De Deyn PP, Marescau B, Qureshi IA. Hyperargininemia: a treatable inborn error of metabolism? In: De Deyn PP, Marescau B, Qureshi IA (eds), Guanidino Compounds, vol.2, J. Libbey, London, 1997. pp. 53-69.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.